

Syntheses, Structures, and Electrochemical Properties of Metallacyclic Oxidovanadium(V) Complexes with Asymmetric Multidentate Linking Ligands


Abstract
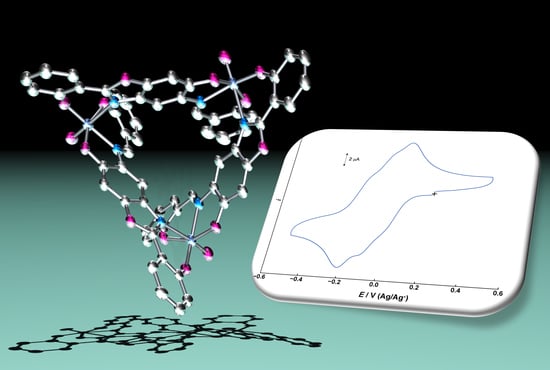
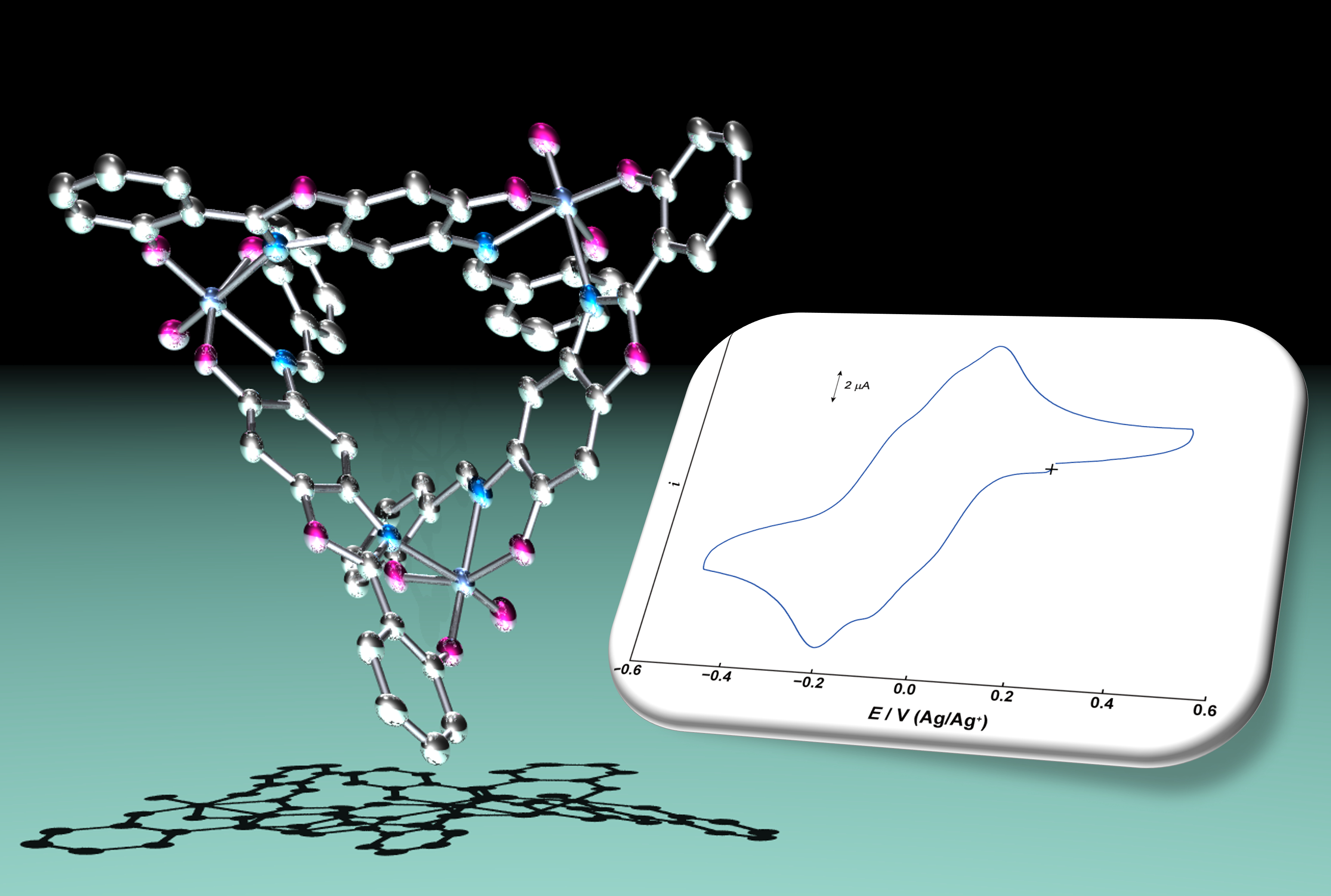
:
1. Introduction
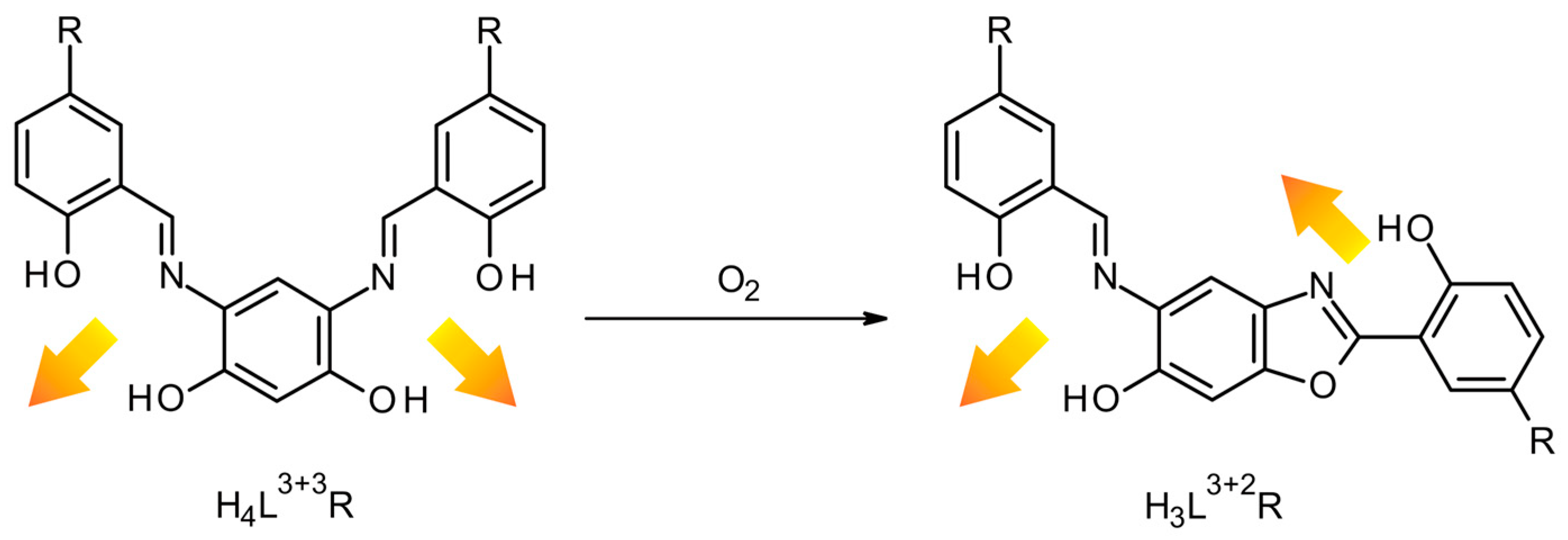
2. Results and Discussion
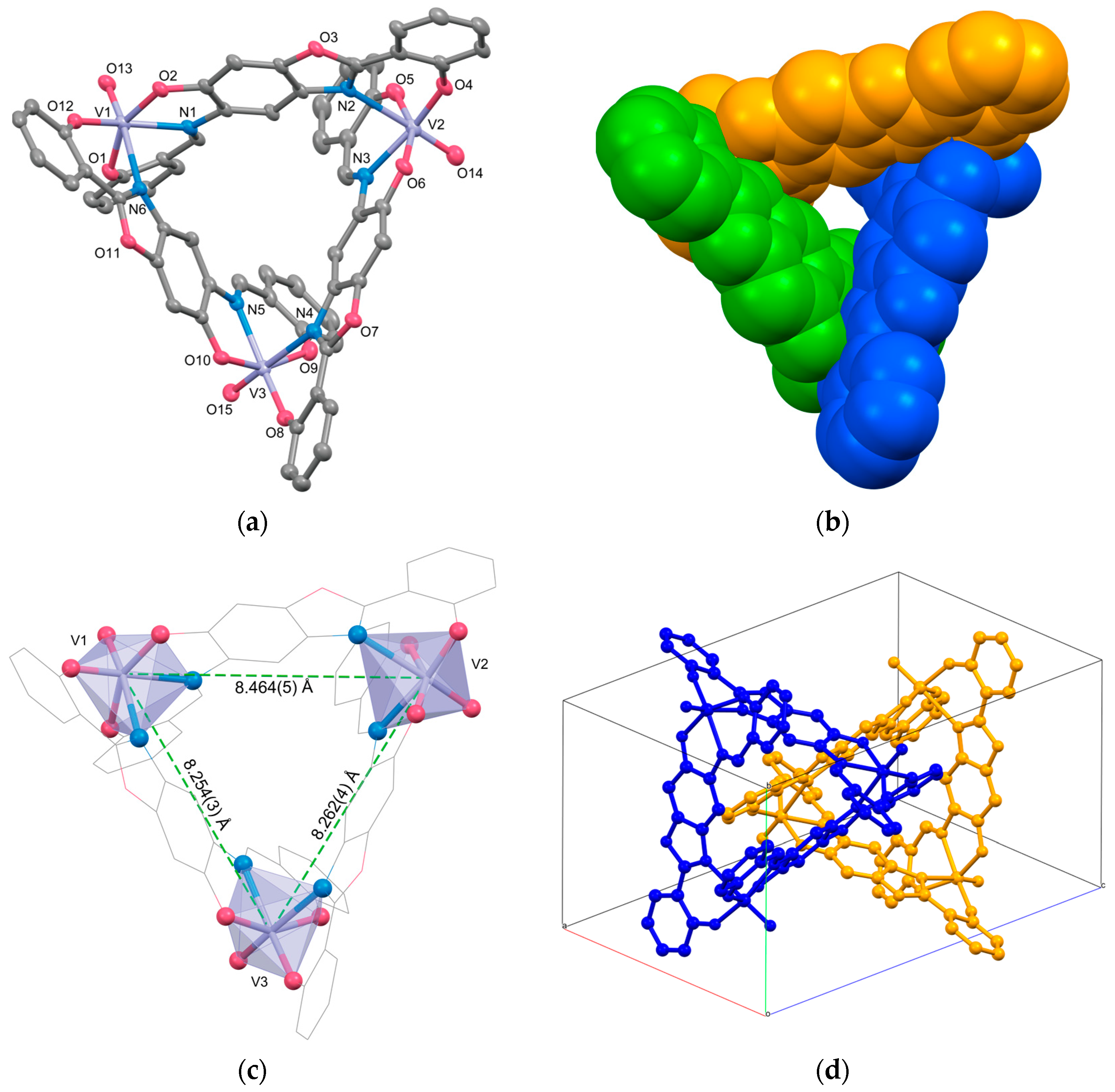
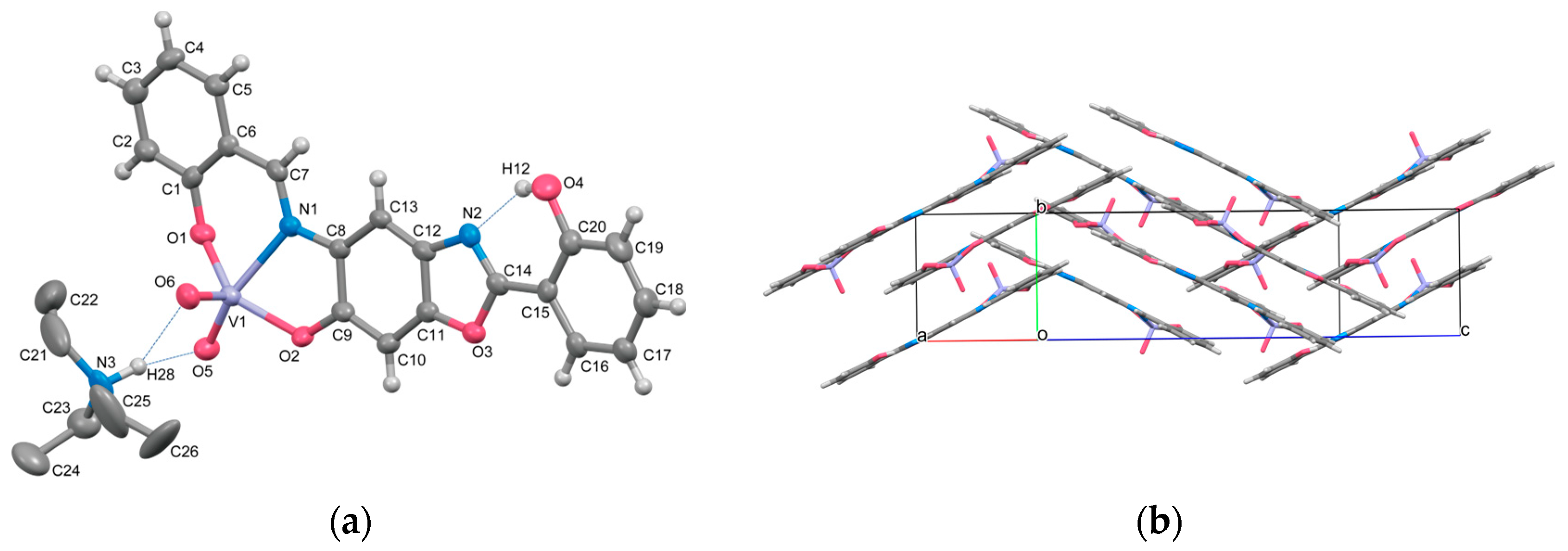
2.1. Crystal Structures
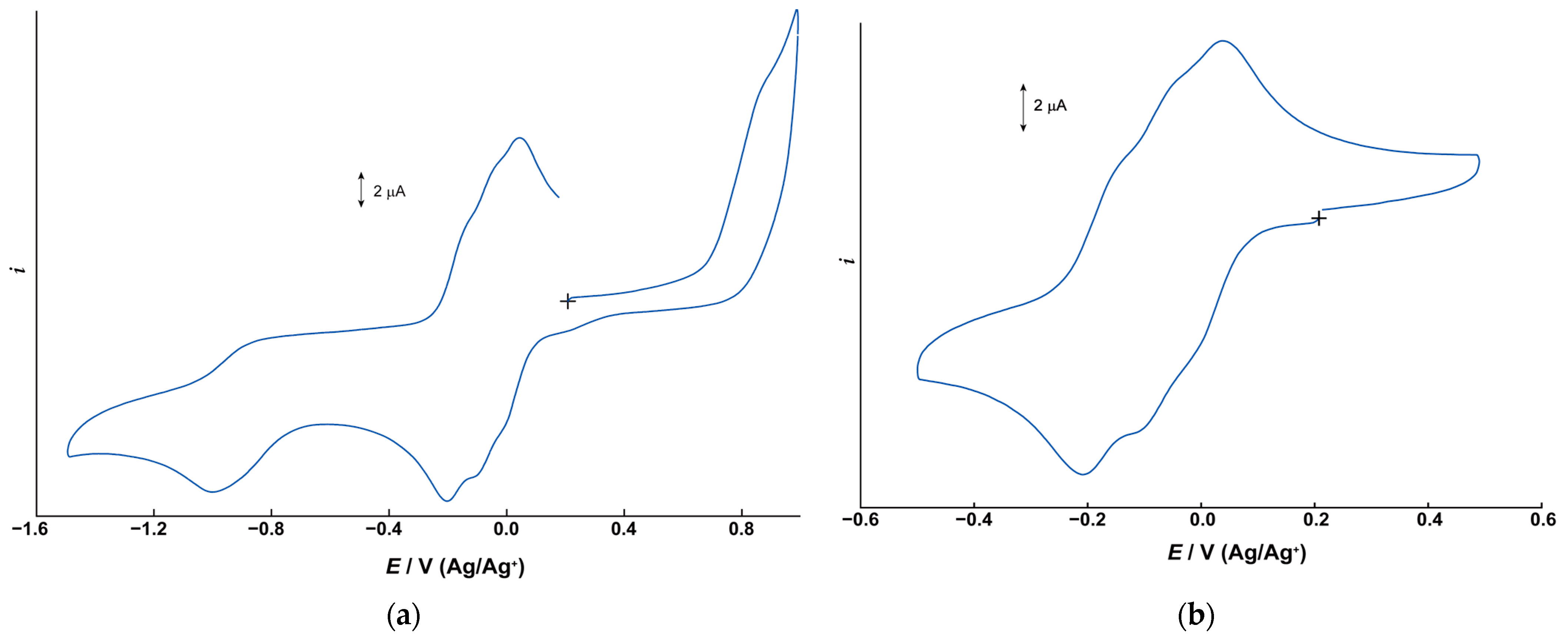
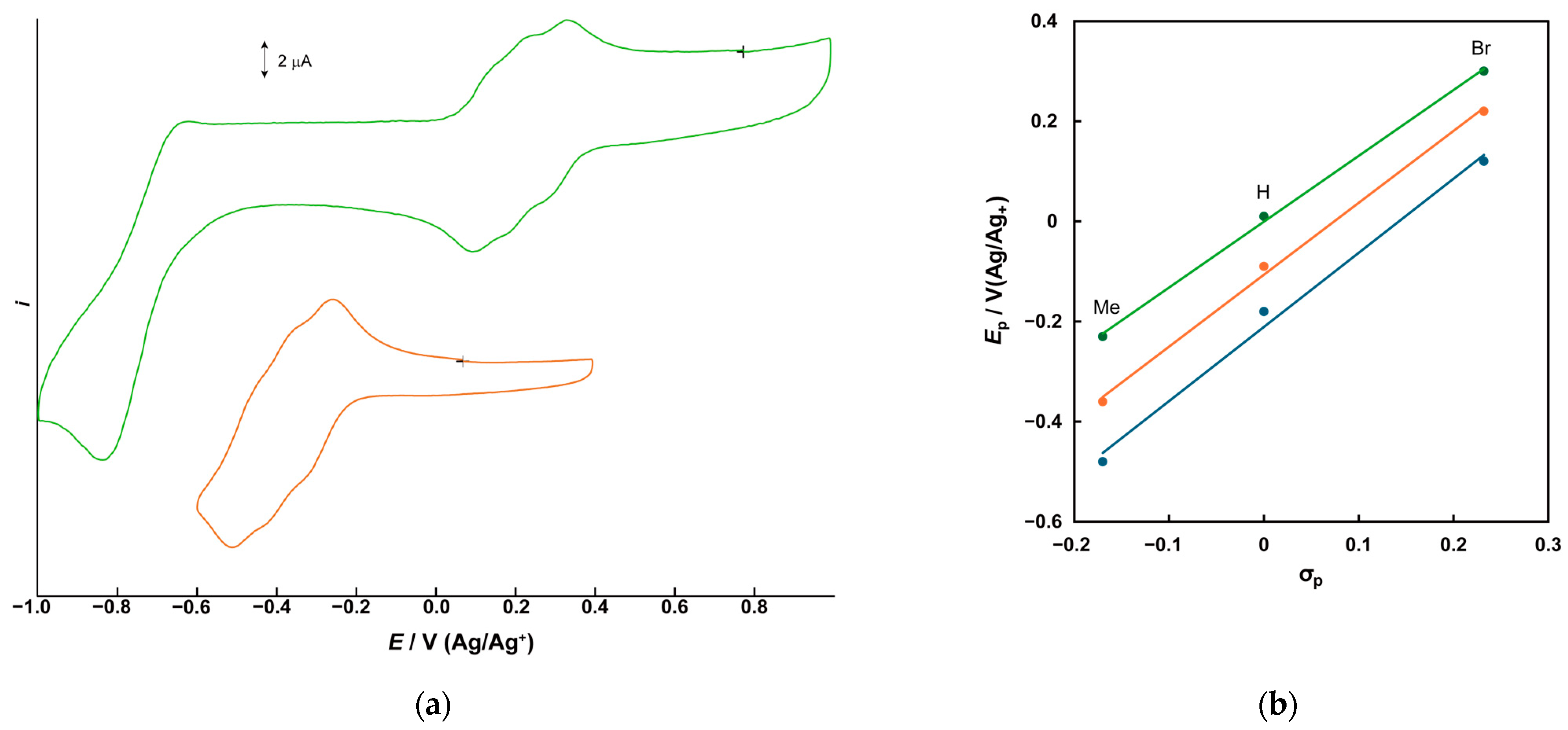
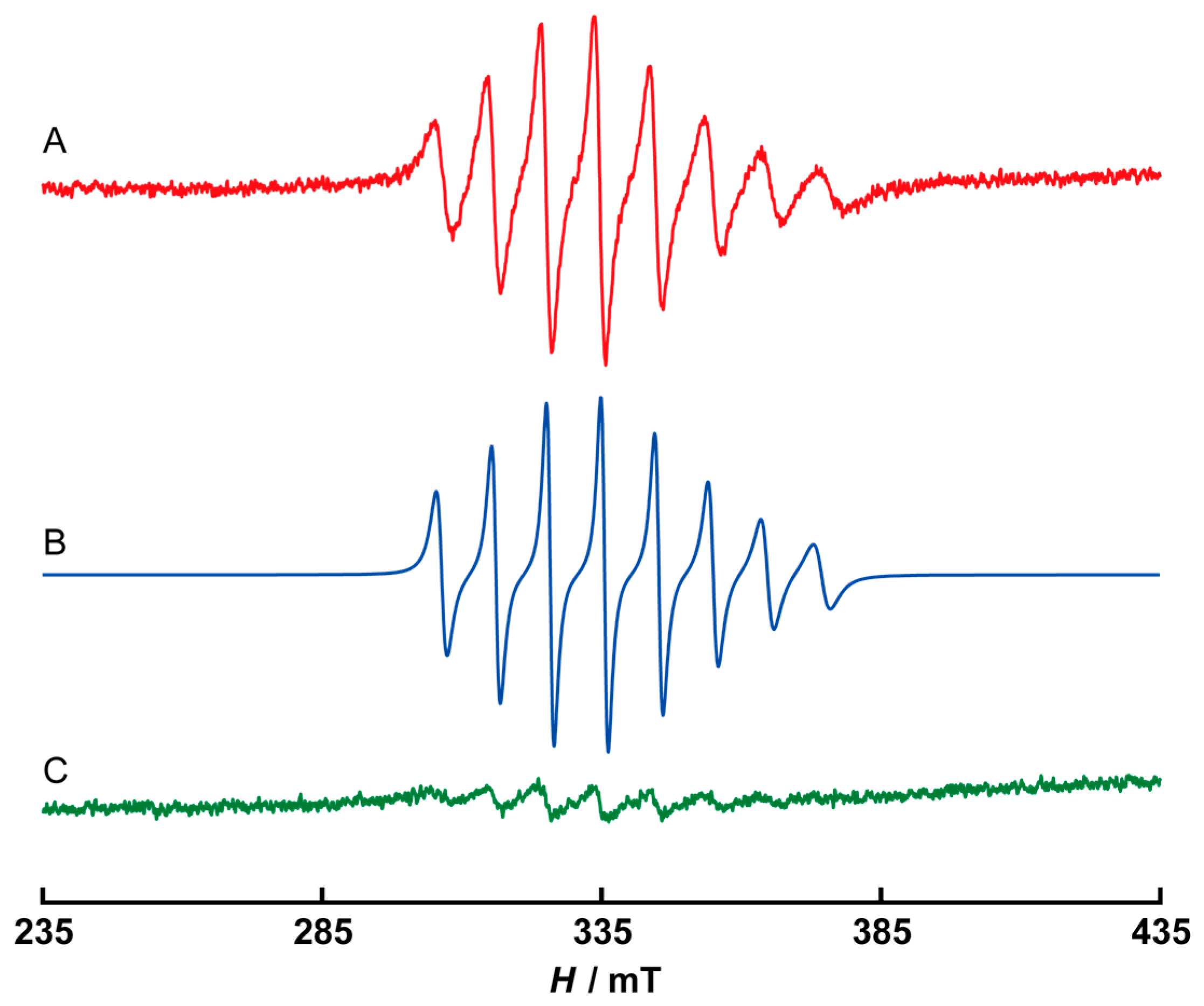
2.2. Electrochemical Properties
3. Materials and Methods
3.1. Materials
3.2. Preparation
3.2.1. Ligands
3.2.2. [{. VO(L3+2R)}3] (R = H: 1, Me: 2, Br: 3)
3.2.3. (. Et3NH)[(VO2)(HL3+2H)] (4)
3.3. Physical Measurements
3.4. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domoto, Y.; Fujita, M. Self-assembly of nanostructures with high complexity based on metal···unsaturated-bond coordination. Coord. Chem. Rev. 2022, 466, 214605. [Google Scholar] [CrossRef]
- Yin, C.; Du, J.; Olenyuk, B.; Stang, P.J.; Sun, Y. The applications of metallacycles and metallacages. Inorganics 2023, 11, 54. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, F.; Shen, X.; He, T.; Qiu, H.; Yin, S.; Stang, P.J. Self-healing metallacycle-cored supramolecular polymers based on a metal−salen complex constructed by orthogonal metal coordination and host−guest interaction with amino acid sensing. ACS Macro Lett. 2021, 10, 873–879. [Google Scholar] [CrossRef]
- Joshi, T.; Graham, B.; Spiccia, L. Macrocyclic metal complexes for metalloenzyme mimicry and sensor development. Acc. Chem. Res. 2015, 48, 2366–2379. [Google Scholar] [CrossRef]
- Swiegers, G.F.; Malefetse, T.J. Classification of coordination polygons and polyhedra according to their mode of self-assembly. 2. Review of the literature. Coord. Chem. Rev. 2002, 225, 91–121. [Google Scholar] [CrossRef]
- Harris, K.; Fujita, D.; Fujita, M. Giant hollow MnL2n spherical complexes: Structure, functionalisation and applications. Chem. Commun. 2013, 49, 6703–6712. [Google Scholar] [CrossRef]
- Elbert, S.M.; Mastalerz, M. Metal salen- and salphen-containing organic polymers: Synthesis and applications. Org. Mater. 2020, 2, 182–203. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional Porous Coordination Polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Zhong, M.; Kong, L.; Zhao, K.; Zhang, Y.-H.; Li, N.; Bu, X.-H. Recent Progress of Nanoscale Metal-Organic Frameworks in Synthesis and Battery Applications. Adv. Sci. 2021, 8, 2001980. [Google Scholar] [CrossRef]
- Jiang, H.; Jin, S.; Wang, C.; Ma, R.; Song, Y.; Gao, M.; Liu, X.; Shen, A.; Cheng, G.J.; Deng, H. Nanoscale Laser Metallurgy and Patterning in Air Using MOFs. J. Am. Chem. Soc. 2019, 141, 5481–5489. [Google Scholar] [CrossRef]
- Muto, M.; Hatae, N.; Tamekuni, Y.; Yamada, Y.; Koikawa, M.; Tokii, T. Tripodal trimanganese(III) complexes of new un-symmetrical pentadentate ligands derived from 2-(salicylideneamino)phenol: Syntheses, crystal structures and properties. Eur. J. Inorg. Chem. 2007, 2007, 3701–3709. [Google Scholar] [CrossRef]
- Manecke, G.; Wille, W.E. Darstellung und eigenschaften chelatbildender monomerer und polymerer Schiffscher basen vom salicylaldehyd und vom 2.5-dihydroxyterephthalaldehyd (teil I). Makromol. Chem. 1970, 133, 61–82. [Google Scholar] [CrossRef]
- Kurusu, T.; Storck, W.; Manecke, G. Die autoxidation von cumol in gegenwart von chelaten monomerer und polymerer Schiffscher basen. Makromol. Chem. 1975, 176, 3185–3200. [Google Scholar] [CrossRef]
- Patra, R.; Mondal, S.; Sinha, D.; Rajak, K.K. Mono Versus Dinuclear Vanadium(V) Complexes: Solvent Dependent Structural Versatility and Electro Syntheses of Mixed-Valence Oxovanadium(IV/V) Entities in Solution. ACS Omega 2022, 7, 11710–11721. [Google Scholar] [CrossRef]
- Borah, R.; Lahkar, S.; Deoriand, N.; Brahma, S. Synthesis, characterization and application of oxovanadium(IV) complexes with [NNO] donor ligands: X-ray structures of their corresponding dioxovanadium(V) complexes. RSC Adv. 2022, 12, 13740–13748. [Google Scholar] [CrossRef]
- Grivani, G.; Delkhosh, S.; Fejfarová, K.; Dušek, M.; Khalaji, A.D. Polynuclear oxovanadium(IV) Schiff base complex [VOL2]n (L = (5-bromo-2- hydroxybenzyl-2-furylmethyl)imine): Synthesis, characterization, crystal structure, catalytic properties and thermal decomposition into V2O5 nano-particles. Inorg. Chem. Commun. 2013, 27, 82–87. [Google Scholar] [CrossRef]
- Mubarak, M.Q.E.; Gérard, E.F.; Blanford, C.F.; Hay, S.; Visser, S.P. How do vanadium chloroperoxidases generate hypochlorite from hydrogen peroxide and chloride? A computational study. ACS Catal. 2020, 10, 14067–14079. [Google Scholar] [CrossRef]
- Wever, R.; Horst, M.A. The role of vanadium haloperoxidases in the formation of volatile brominated compounds and their impact on the environment. Dalton Trans. 2013, 42, 11778–11786. [Google Scholar] [CrossRef]
- Sakurai, H.; Tsuchiya, K. A biomimetic model for vanadium-containing bromoperoxidase. FEBS Lett. 1990, 260, 109–112. [Google Scholar] [CrossRef]
- Berry, R.E.; Armstrong, E.M.; Beddoes, R.L.; Collison, D.; Ertok, S.N.; Helliwell, M.; Garner, C.D. The Structural Characterization of Amavadin. Angew. Chem. Int. Ed. 1999, 15, 795–797. [Google Scholar] [CrossRef]
- Sahu, G.; Tiekink, E.R.T.; Dinda, R. Study of DNA interaction and cytotoxicity activity of oxidovanadium(V) complexes with ONO donor Schiff base ligands. Inorganics 2021, 9, 66. [Google Scholar] [CrossRef]
- Kwiatkowski, E.; Romanowski, G.; Nowicki, W.; Kwiatkowski, M.; Suwińska, K. Dioxovanadium(V) Schiff base complexes of N-methyl-1,2-diaminoethane and 2-methyl-1,2-diaminopropane with aromatic o-hydroxyaldehydes and o-hydroxyketones: Synthesis, characterization, catalytic properties and structure. Polyhedron 2003, 22, 1009–1018. [Google Scholar] [CrossRef]
- Plass, W.; Pohlmann, A.; Yozgatli, H.-P. N-Salicylidenehydrazides as versatile tridentate ligands for dioxovanadium(V) complexes. J. Inorg. Biochem. 2000, 80, 181–183. [Google Scholar] [CrossRef]
- Baruah, B.; Rath, S.P.; Chakravorty, A. A novel pentacoordinated dioxovanadium(V) salicylaldiminate: Solvent specific crystallization of dimorphs with contrasting coordination geometries, ligand conformations and supramolecular architectures. Eur. J. Inorg. Chem. 2004, 2004, 1873–1878. [Google Scholar] [CrossRef]
- Clague, M.J.; Keder, N.L.; Butler, A. Biomimics of vanadium bromoperoxidase: Vanadium(V)-Schiff base catalyzed oxidation of bromide by hydrogen peroxide. Inorg. Chem. 1993, 32, 4754–4761. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[l,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Santis, G.D.; Fabbrizzi, L.; Licchelli, M.; Pallavicini, P. Redox processes in supramolecular coordination compounds. Coord. Chem. Rev. 1992, 120, 237–257. [Google Scholar] [CrossRef]
- Berben, L.A.; Faia, M.C.; Crawford, N.R.M.; Long, J.R. Angle-dependent electronic effects in 4,4′-bipyridine-bridged Ru3 triangle and Ru4 square complexes. Inorg. Chem. 2006, 45, 6378–6386. [Google Scholar] [CrossRef]
- Severin, K. Self-assembled organometallic receptors for small ions. Coord. Chem. Rev. 2003, 245, 3–10. [Google Scholar] [CrossRef]
- Shan, N.; Vickers, S.J.; Adams, H.; Ward, M.D.; Thomas, J.A. Switchable electron-transfer processes in a mixed-valence, kinetically locked, trinuclear RuII metallamacrocycle. Angew. Chem. Int. Ed. 2004, 43, 3938–3941. [Google Scholar] [CrossRef]
- Galloni, P.; Coletti, A.; Floris, B.; Conte, V. Electrochemical properties of VO salen complexes. Inorg. Chim. Acta 2014, 420, 144–148. [Google Scholar] [CrossRef]
- Lanznaster, M.; Neves, A.; Bortoluzzi, A.J.; Assumpção, A.M.C.; Vencato, I.; Machado, S.P.; Drechsel, S.M. Electronic effects of electron-donating and -withdrawing groups in model complexes for iron-tyrosine-containing metalloenzymes. Inorg. Chem. 2006, 45, 1005–1011. [Google Scholar] [CrossRef]
- Koikawa, M.; Ōkawa, H.; Kida, S. Manganese (IV) and manganese (V) complexes with N-(2-hydroxyphenyl)salicylamides. J. Chem. Soc. Dalton Trans. 1988, 641–645. [Google Scholar] [CrossRef]
- Koikawa, M. HyperFine—ESR Simulator; Version 2.8; Saga University: Saga, Japan, 2023. [Google Scholar]
- Tasiopoulos, A.J.; Troganis, A.N.; Evangelou, A.; Raptopoulou, C.P.; Terzis, A.; Deligiannakis, Y.; Kabanos, T.A. Synthetic analogues for oxovanadium(IV)–glutathione interaction: An EPR, synthetic and structural study of oxovanadium(IV) compounds with sulfhydryl-containing pseudopeptides and dipeptides. Chem. Eur. J. 1999, 5, 910–921. [Google Scholar] [CrossRef]
- Ando, R.; Ono, H.; Yagyu, T.; Maeda, M. Spectroscopic characterization of mononuclear, binuclear, and insoluble polynuclear oxovanadium(IV)–Schiff base complexes and their oxidation catalysis. Inorg. Chim. Acta 2004, 357, 817–823. [Google Scholar] [CrossRef]
- Matsunaga, Y. ESR spectra of bis(acetylacetonato)oxovanadium(IV) and related chelates in molten o-terphenyl and some other organic compounds. Bull. Chem. Soc. Jpn. 1978, 5, 422–426. [Google Scholar] [CrossRef]
- Kaimal, V.R.M.; Shome, S.C. Direct titration of vanadium(IV) with EDTA using N-benzoyl-N-phenylhydroxylamine as metal indicator. Anal. Chim. Acta 1962, 27, 594–596. [Google Scholar] [CrossRef]
- Rigaku Corporation. CrystalClear: Data Collection and Processing Software; Rigaku Corporation: Tokyo, Japan, 1998. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. 2015, C71, 9–18. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomano, V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.; Polidori, G.; Camalli, M. SIRPOW.92—A program for automatic solution of crystal structures by direct methods optimized for powder data. J. Appl. Cryst. 1994, 27, 435–436. [Google Scholar] [CrossRef]
- Rigaku Corporation. CrystalStructure: Crystal Structure Analysis Package (Version 4.3); Rigaku Corporation: Tokyo, Japan, 2000. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 4 | |
|---|---|---|
| Empirical formula | C60H33N6O15V3 | C26H28N3O6V |
| Formula weight | 1230.74 | 529.47 |
| Temperature/K | 113 | 301 |
| Crystal system | triclinic | Monoclinic |
| Space group | P | P21/n |
| a/Å | 12.465(5) | 11.978(3) |
| b/Å | 16.244(6) | 7.5880(14) |
| c/Å | 18.285(8) | 28.042(5) |
| α/° | 102.99(3) | 90 |
| β/° | 98.16(3) | 101.705(18) |
| γ/° | 108.59(3) | 90 |
| V/Å3 | 3327(3) | 2495.7(8) |
| Z | 2 | 4 |
| Dcalc/g cm−3 | 1.229 | 1.409 |
| μ(MoKα)/mm−1 | 0.476 | 0.444 |
| F(0 0 0) | 1248.0 | 1104.0 |
| Crystal dimensions/mm3 | 0.22 × 0.14 × 0.08 | 0.40 × 0.20 × 0.10 |
| Radiation | MoKα (λ = 0.71075) | MoKα (λ = 0.71075) |
| 2θ range for data collection/° | 6.1 to 54.838 | 6.0 to 55.0 |
| No. of reflections collected | 26,785 | 5577 |
| No. of independent reflections | 14,423 | 5478 |
| Data/Restraints/Params | 144,23/0/758 | 5478/0/329 |
| Goodness of fit indicator | 0.949 | 0.947 |
| R indices [I > 2.00σ(I)] | R1 = 0.0568 | R1 = 0.0790 |
| wR2 = 0.1509 | wR2 = 0.2160 | |
| R indices (all data) | R1 = 0.0852 | R1 = 0.2748 |
| wR2 = 0.1766 | wR2 = 0.3181 | |
| Largest diff. peak, hole/e Å−3 | 0.41, −0.61 | 0.64, −0.60 |
| CCDC deposition number | 2,338,377 | 2,338,378 |
| Bond | Distance/Å | Angle | Angle/° |
|---|---|---|---|
| V–O1 | 1.897(2) | O1–V1–O2 | 154.33(9) |
| V1–O2 | 1.944(2) | O12–V1–N1 | 162.90(10) |
| V1–O12 | 1.847(3) | O13–V1–N6 | 172.12(10) |
| V1–O13 | 1.601(2) | O12–V1–N6 | 79.98(9) |
| V1–N1 | 2.121(3) | O1–V1–N1 | 83.27(10) |
| V1–N6 | 2.355(3) | O2–V1–N1 | 77.24(10) |
| V2–O4 | 1.820(2) | O5–V2–O6 | 156.00(10) |
| V2–O5 | 1.862(2) | O4–V2–N3 | 164.98(11) |
| V2–O6 | 1.941(2) | O14–V2–N2 | 177.48(10) |
| V2–O14 | 1.609(3) | O4–V2–N2 | 81.01(10) |
| V2–N2 | 2.436(3) | O5–V2–N3 | 85.17(10) |
| V2–N3 | 2.110(3) | O6–V2–N3 | 78.29(10) |
| V3–O8 | 1.838(2) | O9–V3–O10 | 154.56(9) |
| V3–O9 | 1.893(3) | O8–V3–N5 | 162.18(11) |
| V3–O10 | 1.943(2) | O15–V3–N4 | 174.62(12) |
| V3–O15 | 1.606(2) | O8–V3–N4 | 80.91(10) |
| V3–N4 | 2.312(3) | O9–V3–N5 | 83.70(10) |
| V3–N5 | 2.099(3) | O10–V3–N5 | 77.88(10) |
| V1⋯V2 | 8.464(5) | ||
| V1⋯V3 | 8.254(3) | ||
| V2⋯V3 | 8.262(4) |
| Bond | Distance/Å | Angle | Angle/° |
|---|---|---|---|
| V1–O1 | 1.884(6) | O1–V1–O2 | 140.9(2) |
| V1–O2 | 1.941(6) | O1–V1–O5 | 95.8(2) |
| V1–O5 | 1.644(5) | O1–V1–O6 | 105.2(3) |
| V1–O6 | 1.632(6) | O1–V1–N1 | 81.6(2) |
| V1–N1 | 2.207(6) | O2–V1–O5 | 93.6(2) |
| O2–V1–O6 | 108.5(3) | ||
| O4–H12⋯N2 | 2.660(9) | O2–V1–N1 | 75.5(2) |
| N3–H28⋯O5 | 2.900(9) | O5–V1–O6 | 105.8(3) |
| N3–H28⋯O6 | 2.983(9) | O5–V1–N1 | 157.3(3) |
| O6–V1–N1 | 96.6(2) |
| Complex | V3(IV,IV,IV)/V3(IV,IV,V) * | V3(IV,IV,V)/V3(IV,V,V) * | V3(IV,V,V)/V3(V,V,V) * | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Epc | Epa | E1/2 | Epc | Epa | E1/2 | Epc | Epa | E1/2 | |
| 1 | −0.208 | −0.15 ** | −0.18 | −0.12 ** | −0.06 ** | −0.09 | −0.02 ** | +0.038 | +0.01 |
| 2 | −0.512 | −0.44 ** | −0.48 | −0.39 ** | −0.32 ** | −0.36 | −0.20 ** | −0.262 | −0.23 |
| 3 | +0.091 | +0.15 ** | +0.12 | +0.18 ** | +0.25 ** | +0.22 | +0.27 ** | +0.333 | +0.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, K.; Muto, M.; Hamada, M.; Yamada, Y.; Tokii, T.; Koikawa, M. Syntheses, Structures, and Electrochemical Properties of Metallacyclic Oxidovanadium(V) Complexes with Asymmetric Multidentate Linking Ligands. Molecules 2024, 29, 1700. https://doi.org/10.3390/molecules29081700
Hasegawa K, Muto M, Hamada M, Yamada Y, Tokii T, Koikawa M. Syntheses, Structures, and Electrochemical Properties of Metallacyclic Oxidovanadium(V) Complexes with Asymmetric Multidentate Linking Ligands. Molecules. 2024; 29(8):1700. https://doi.org/10.3390/molecules29081700
Chicago/Turabian StyleHasegawa, Kyoko, Masahiro Muto, Masanobu Hamada, Yasunori Yamada, Tadashi Tokii, and Masayuki Koikawa. 2024. "Syntheses, Structures, and Electrochemical Properties of Metallacyclic Oxidovanadium(V) Complexes with Asymmetric Multidentate Linking Ligands" Molecules 29, no. 8: 1700. https://doi.org/10.3390/molecules29081700
APA StyleHasegawa, K., Muto, M., Hamada, M., Yamada, Y., Tokii, T., & Koikawa, M. (2024). Syntheses, Structures, and Electrochemical Properties of Metallacyclic Oxidovanadium(V) Complexes with Asymmetric Multidentate Linking Ligands. Molecules, 29(8), 1700. https://doi.org/10.3390/molecules29081700