
Experimental and Theoretical Insights into the Intermolecular Interactions in Saturated Systems of Dapsone in Conventional and Deep Eutectic Solvents




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
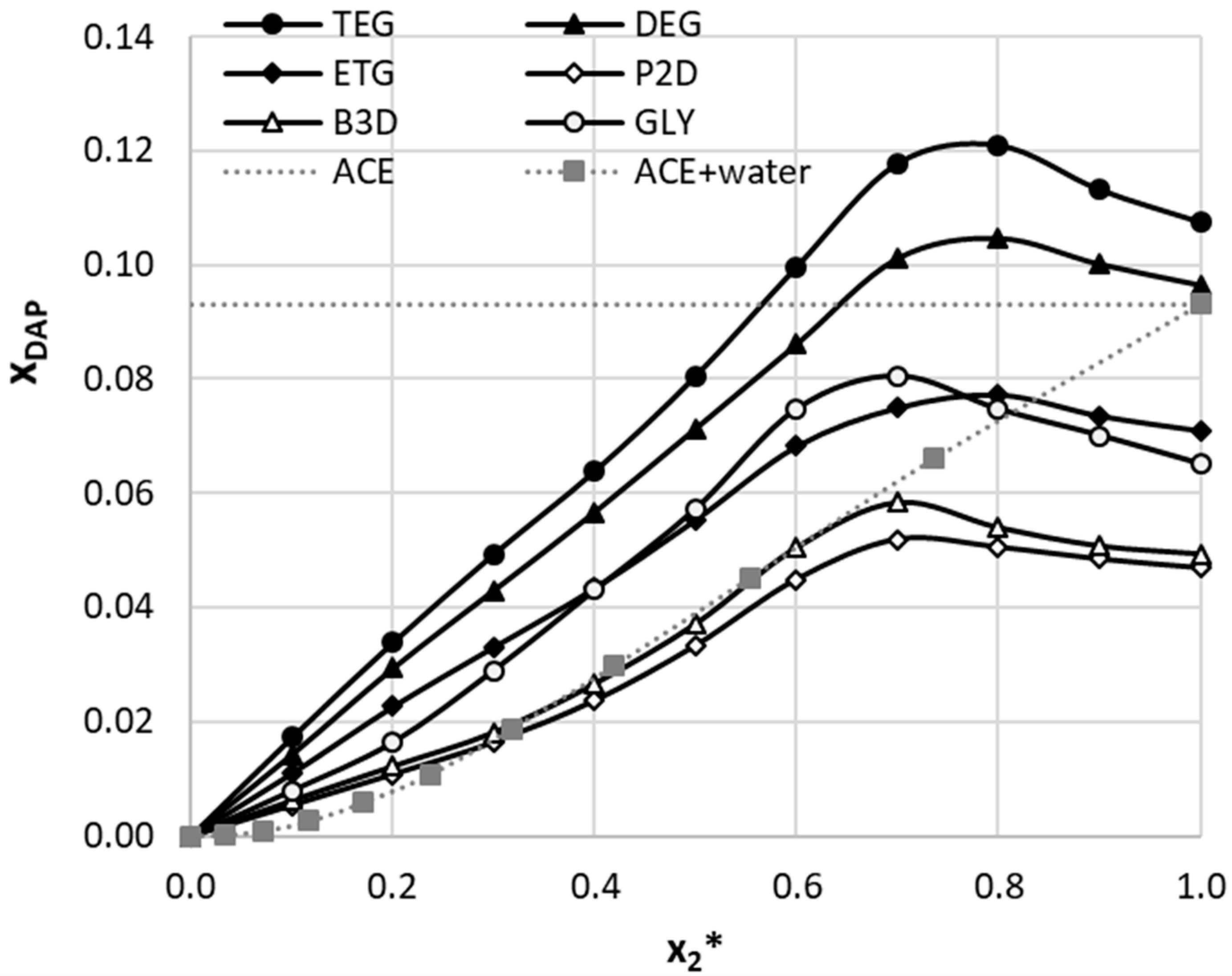
2.1. Solubility of Dapsone in Deep Eutectic Solvents
2.2. Dapsone Solubility Dataset
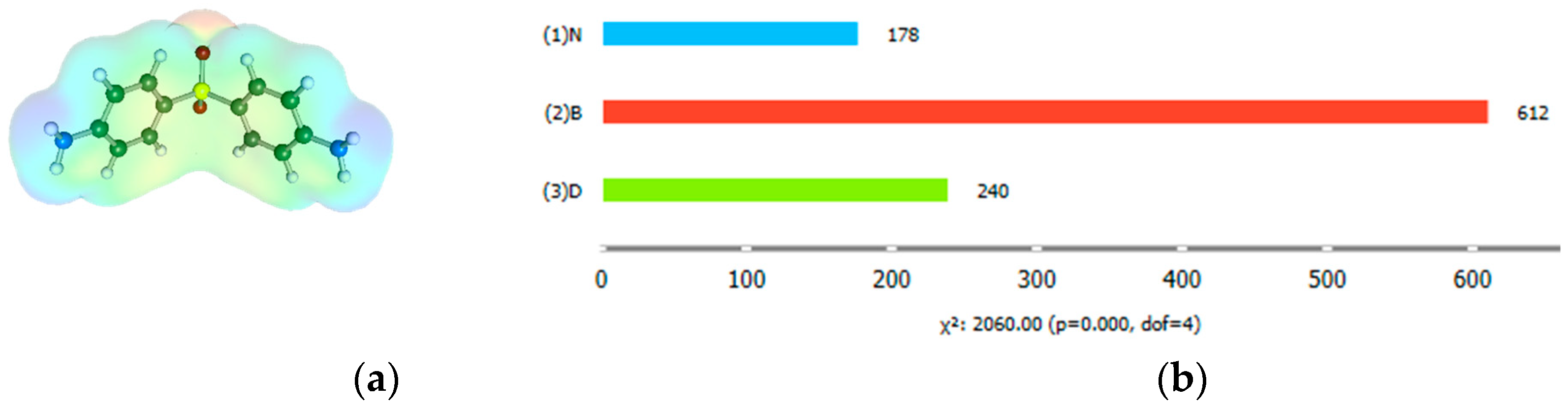
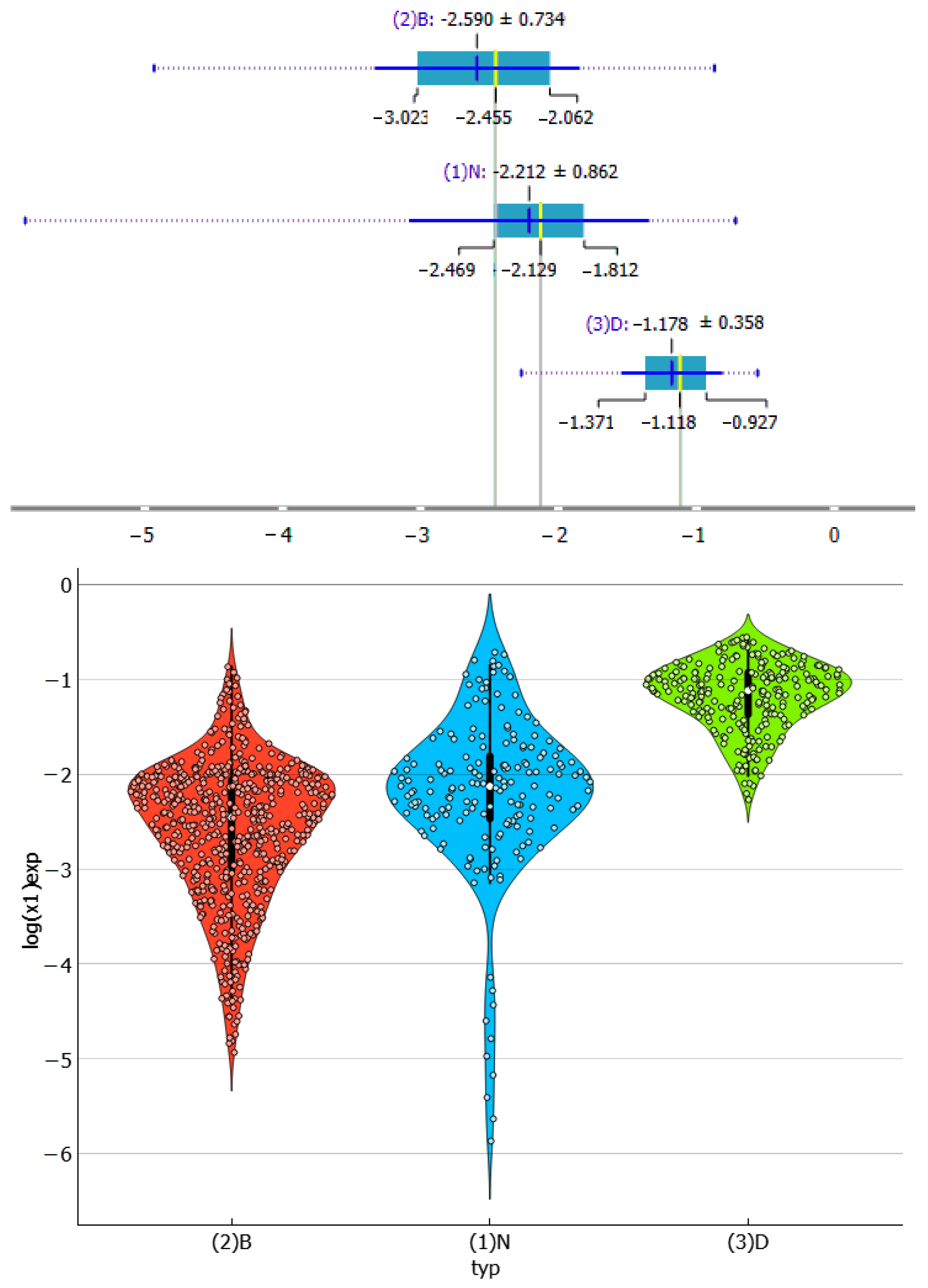
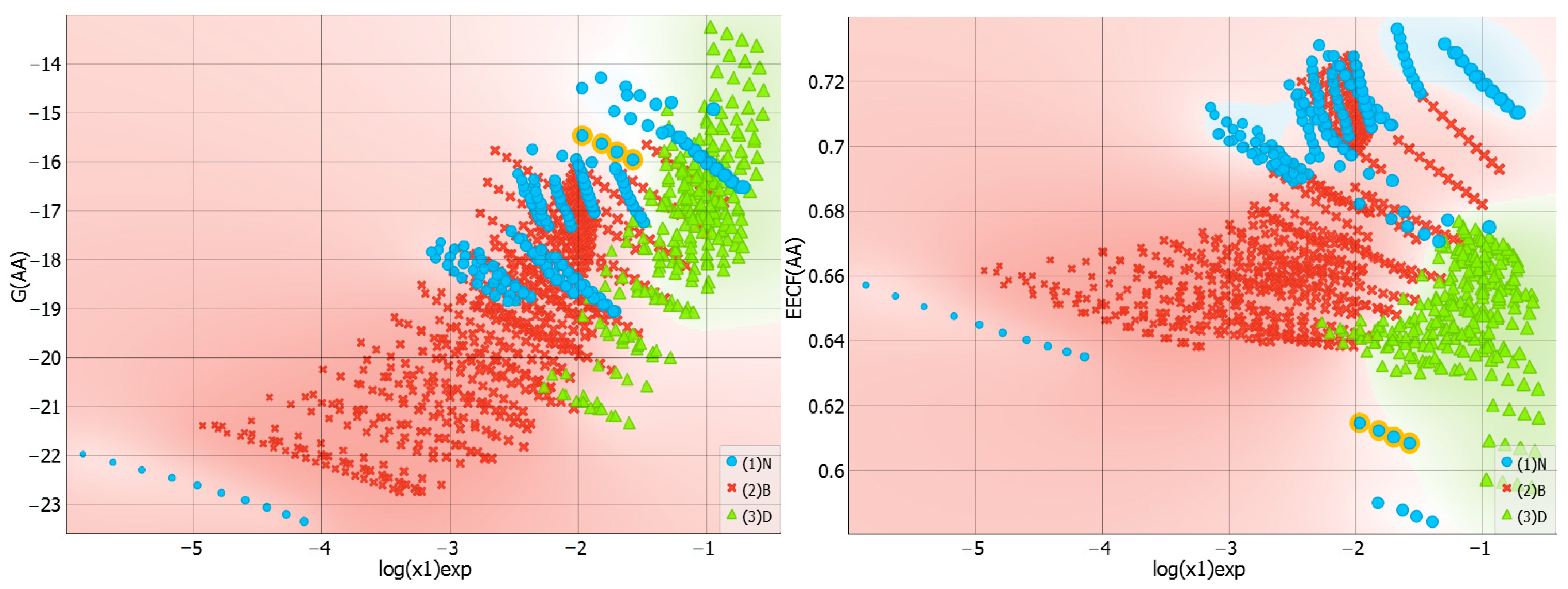
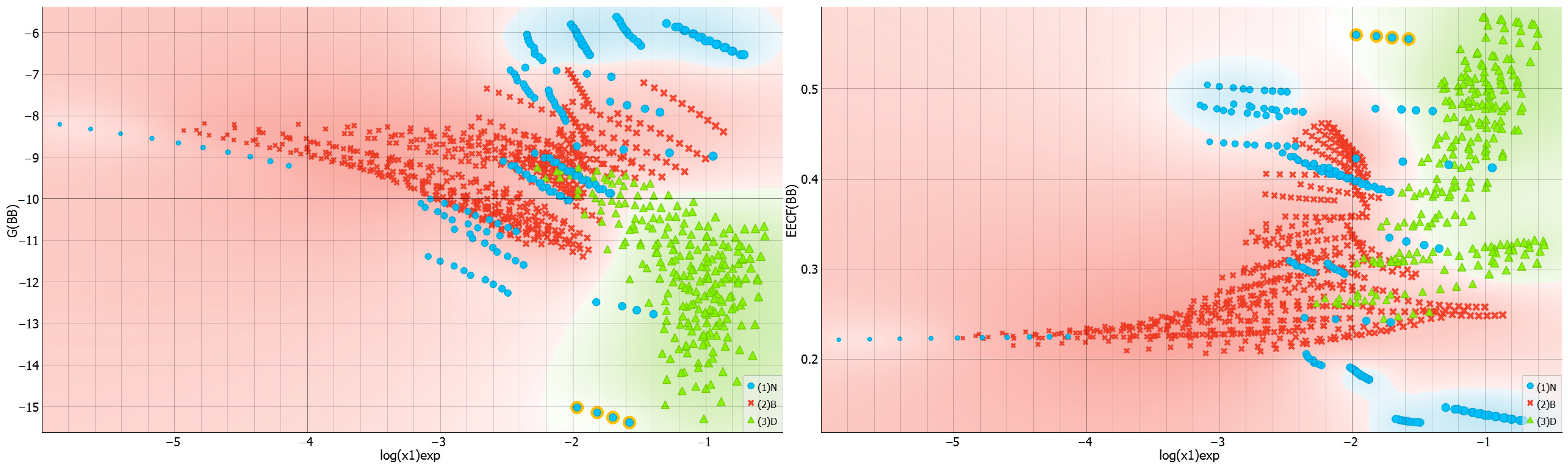
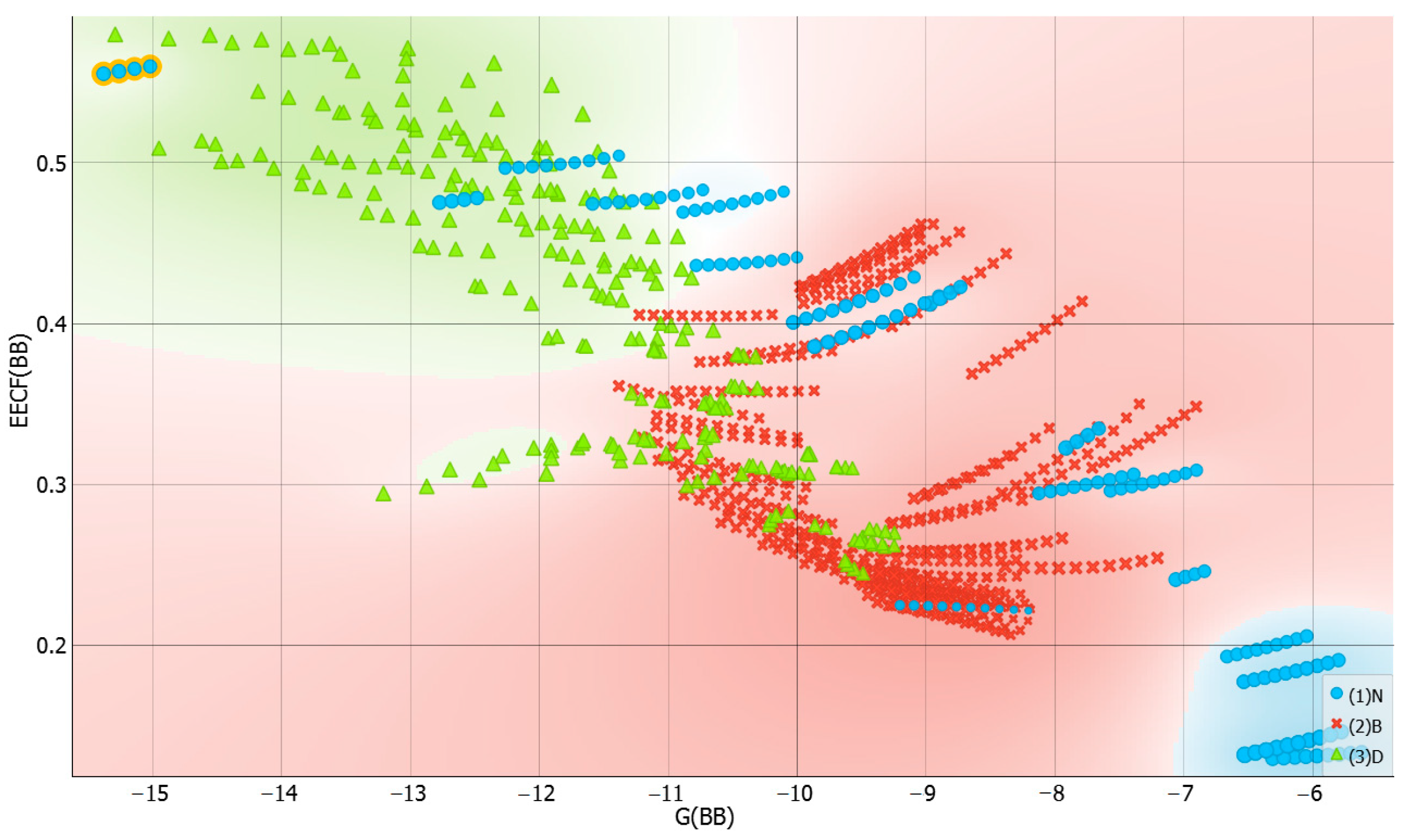
2.3. Distributions of Solute–Solute Affinity in Saturated Systems
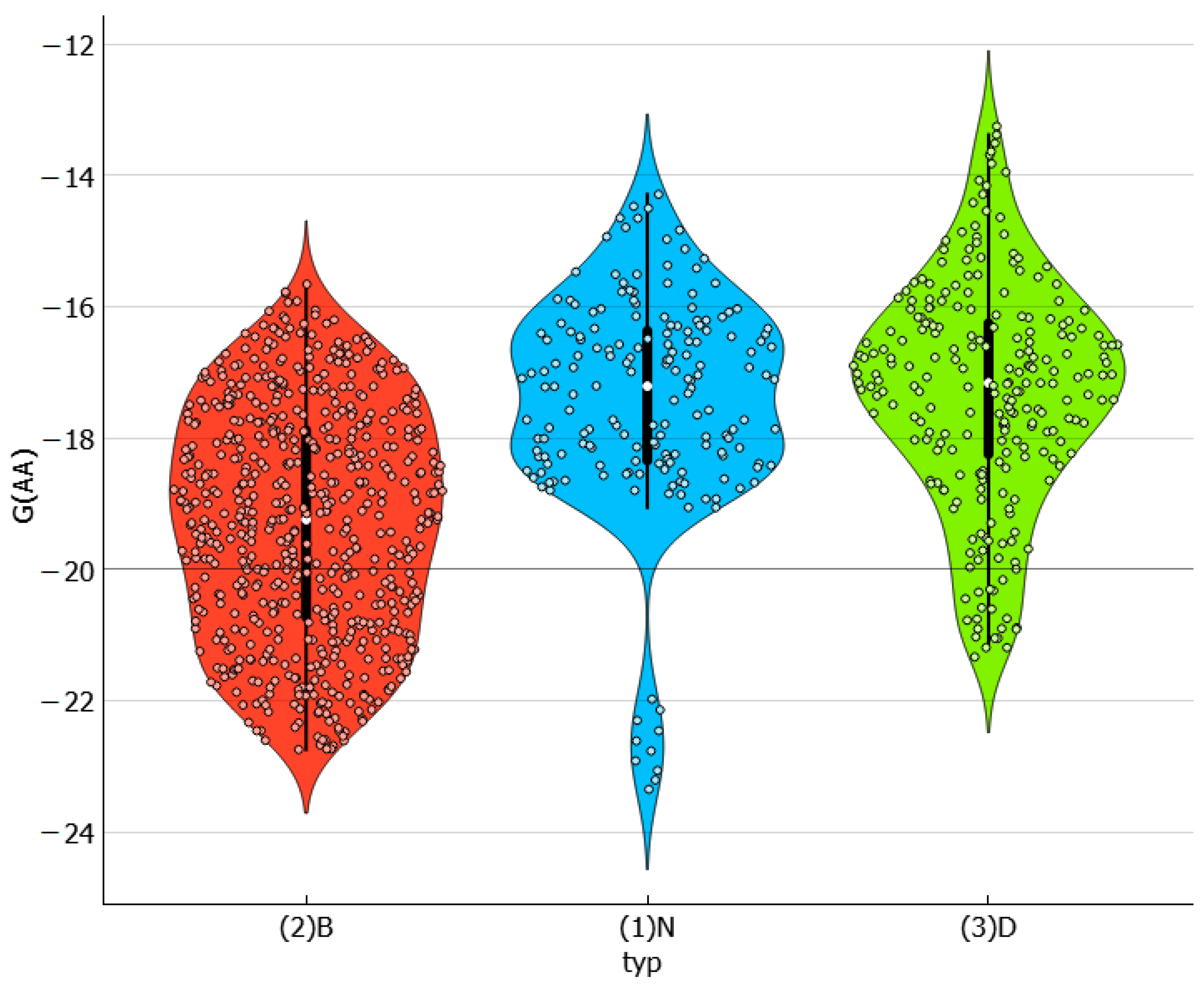
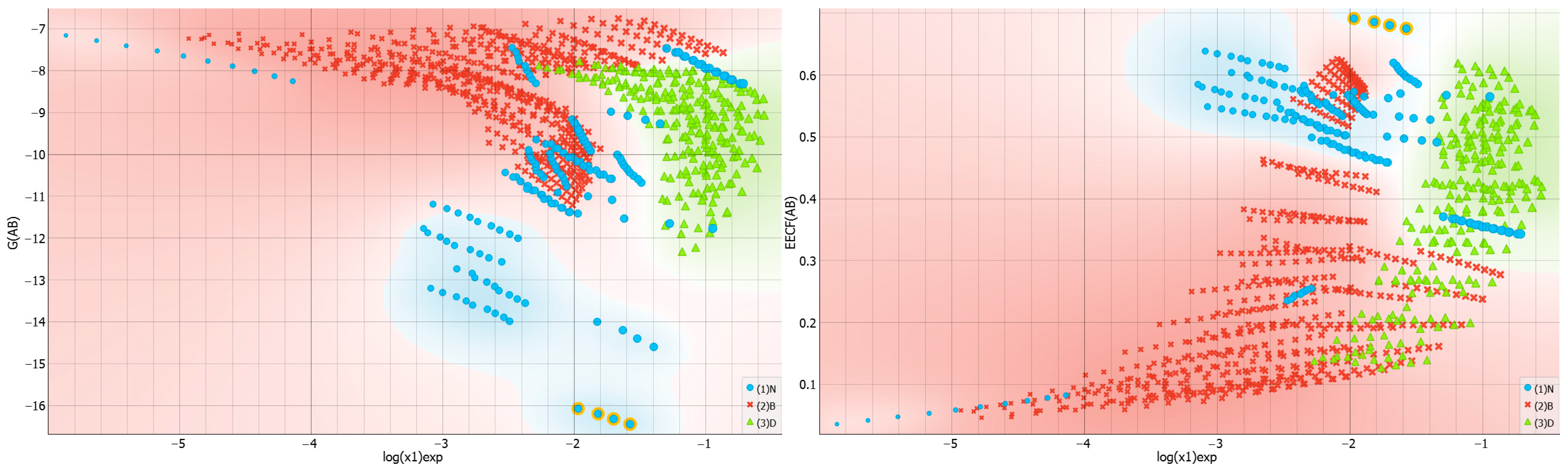
2.4. Distributions of Solute–Solvent Affinity in Saturated Systems
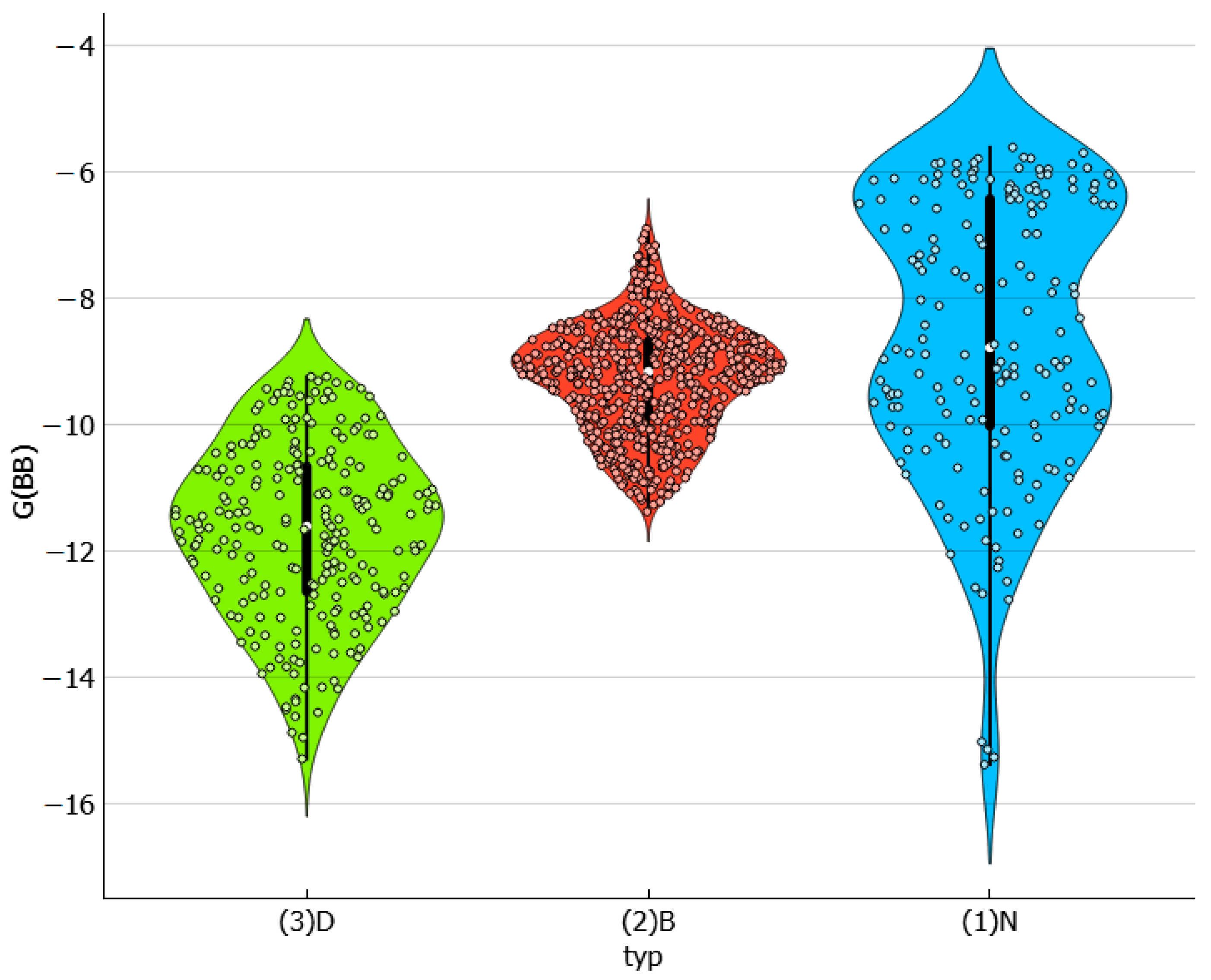
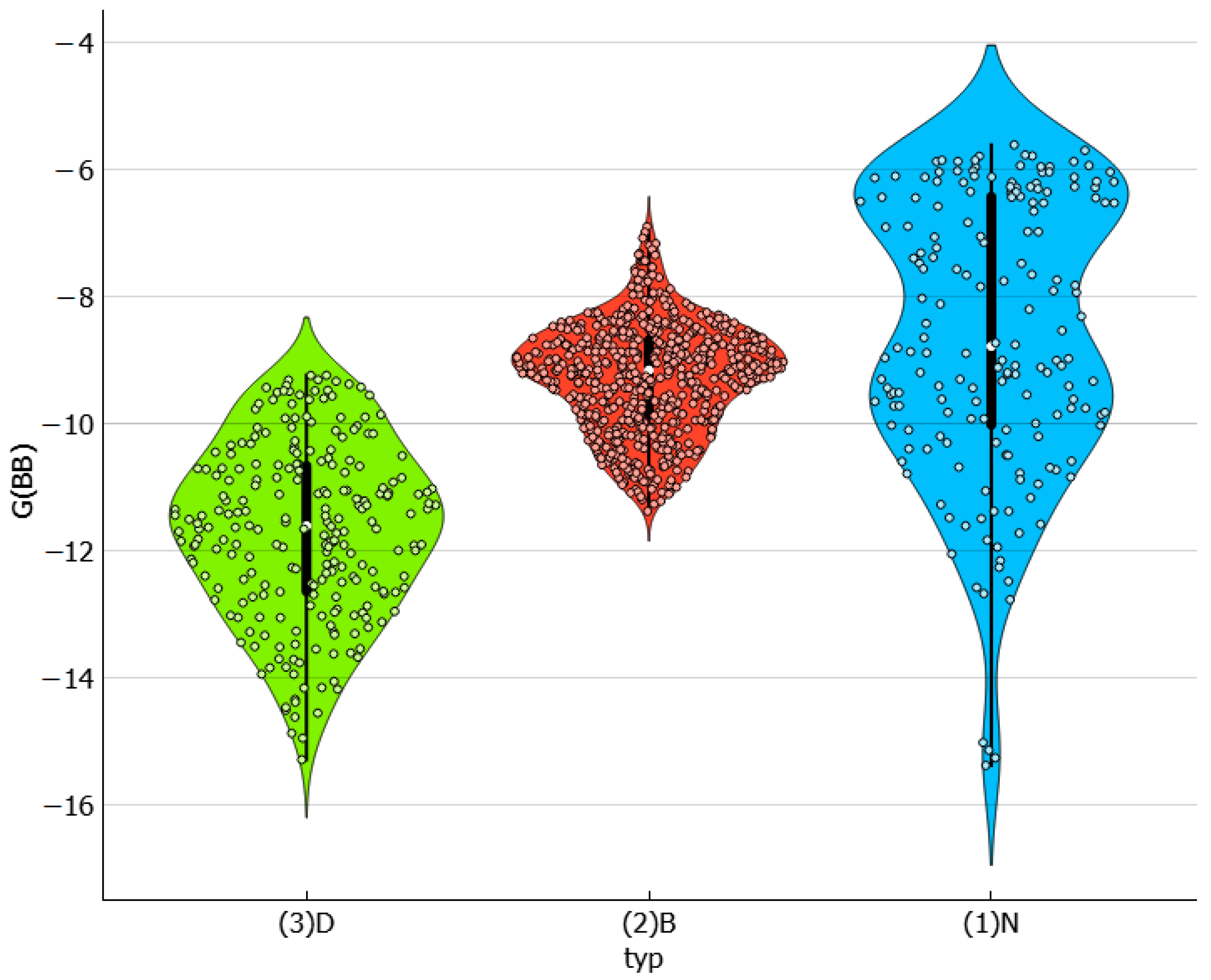
2.5. Solvent–Solvent Interactions in Saturated Systems
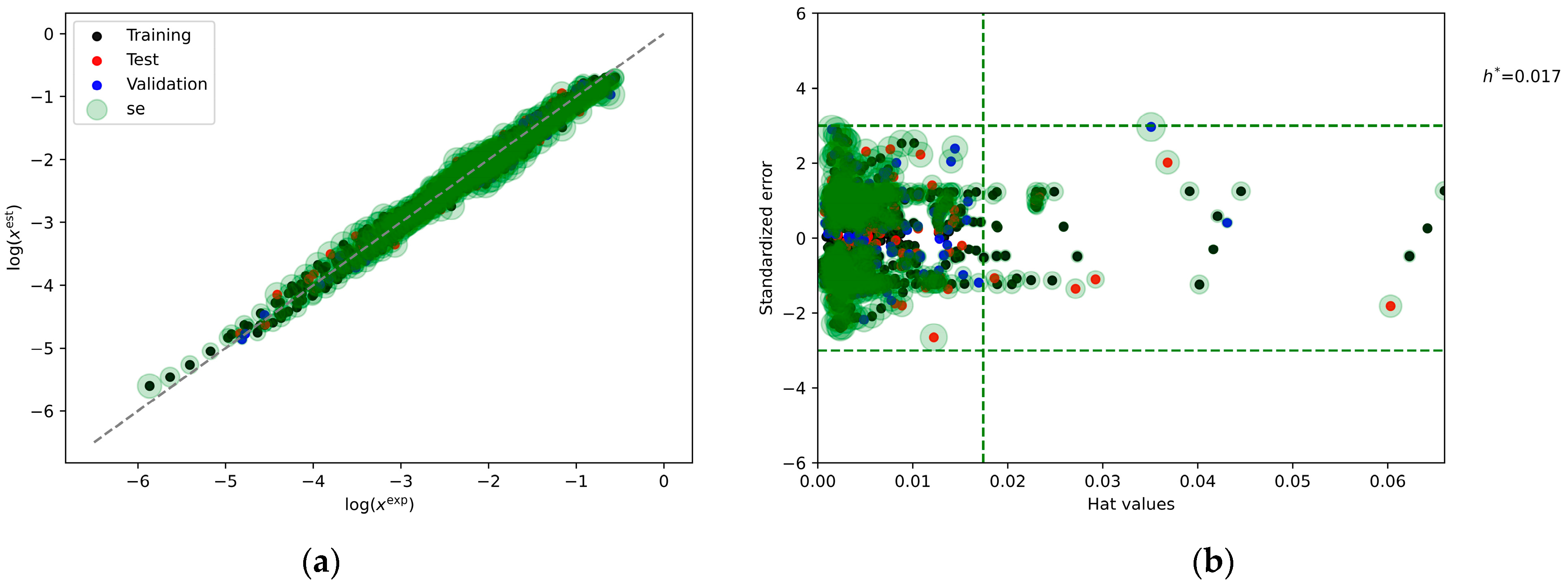
2.6. Machine Learning Models
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Preparation of the Samples and Solubility Measurements
3.2.2. Intermolecular Interaction Quantification
3.2.3. Conformational Analysis
3.2.4. Conformational Search for Pairs Structures
3.2.5. Pattern Identification with Orange Data-Mining
3.2.6. Machine Learning Modelling
- Regularization parameter (C) that controls the trade-off between achieving a smooth decision boundary and fitting the training data;
- Degree for polynomial kernel;
- Gamma defining the influence of a single training example and affects the shape of the decision boundary;
- nu parameter, which is specific to NuSVR and represents an upper bound on the fraction of margin errors and a lower bound on the fraction of support vectors. It is a crucial parameter that influences the model’s flexibility and generalization.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolf, R.; Matz, H.; Orion, E.; Tuzun, B.; Tuzun, Y. Dapsone. Dermatol. Online J. 2002, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Madanipour, M.R.; Fatehi-zardalou, M.; Rahimi, N.; Hemmati, S.; Alaeddini, M.; Etemad-Moghadam, S.; Shayan, M.; Dabiri, S.; Dehpour, A.R. The anti-inflammatory effect of dapsone on ovalbumin-induced allergic rhinitis in balb/c mice. Life Sci. 2022, 297, 120449. [Google Scholar] [CrossRef]
- Zhu, Y.I.; Stiller, M.J. Dapsone and sulfones in dermatology: Overview and update. J. Am. Acad. Dermatol. 2001, 45, 420–434. [Google Scholar] [CrossRef]
- May, S.M.; Motosue, M.S.; Park, M.A. Dapsone is often tolerated in HIV-infected patients with history of sulfonamide antibiotic intolerance. J. Allergy Clin. Immunol. Pract. 2017, 5, 831–833. [Google Scholar] [CrossRef]
- Wozel, G.; Blasum, C. Dapsone in dermatology and beyond. Arch. Dermatol. Res. 2014, 306, 103. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Calvo, A.; Schwartz, J.; Navarro-Blasco, I.; González-Peñas, E.; Sanmartín, C.; Irache, J.; Espuelas, S. Evaluation of Skin Permeation and Retention of Topical Dapsone in Murine Cutaneous Leishmaniasis Lesions. Pharmaceutics 2019, 11, 607. [Google Scholar] [CrossRef]
- Sharquie, K.E.; Najim, R.A.; Abu-Raghif, A.R. Dapsone in Behçet’s Disease: A Double-Blind, Placebo-Controlled, Cross-Over Study. J. Dermatol. 2002, 29, 267–279. [Google Scholar] [CrossRef]
- Chang, D.J.; Lamothe, M.; Stevens, R.M.; Sigal, L.H. Dapsone in rheumatoid arthritis. Semin. Arthritis Rheum. 1996, 25, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.S.; Paidesetty, S.K.; Dehury, B.; Sahoo, J.; Vedithi, S.C.; Mahapatra, N.; Hussain, T.; Padhy, R.N. Molecular docking and simulation study for synthesis of alternative dapsone derivative as a newer antileprosy drug in multidrug therapy. J. Cell. Biochem. 2018, 119, 9838–9852. [Google Scholar] [CrossRef]
- Roman, C.; Dima, B.; Muyshont, L.; Schurmans, T.; Gilliaux, O. Indications and efficiency of dapsone in IgA vasculitis (Henoch-Schonlein purpura): Case series and a review of the literature. Eur. J. Pediatr. 2019, 178, 1275–1281. [Google Scholar] [CrossRef]
- Ghaoui, N.; Hanna, E.; Abbas, O.; Kibbi, A.G.; Kurban, M. Update on the use of dapsone in dermatology. Int. J. Dermatol. 2020, 59, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ríos, C.; Orozco-Suarez, S.; Salgado-Ceballos, H.; Mendez-Armenta, M.; Nava-Ruiz, C.; Santander, I.; Barón-Flores, V.; Caram-Salas, N.; Diaz-Ruiz, A. Anti-Apoptotic Effects of Dapsone after Spinal Cord Injury in Rats. Neurochem. Res. 2015, 40, 1243–1251. [Google Scholar] [CrossRef]
- Zhang, T.; Tian, X.; Wang, Q.; Tong, Y.; Wang, H.; Li, Z.; Li, L.; Zhou, T.; Zhan, R.; Zhao, L.; et al. Surgical stress induced depressive and anxiety like behavior are improved by dapsone via modulating NADPH oxidase level. Neurosci. Lett. 2015, 585, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Tingle, M.; Mahmud, R.; Maggs, J.; Pirmohamed, M.; Park, B. Comparison of the metabolism and toxicity of dapsone in rat, mouse and man. J. Pharmacol. Exp. Ther. 1997, 283, 817–823. [Google Scholar] [PubMed]
- Mitra, A.K.; Thummel, K.E.; Kalhorn, T.F.; Kharasch, E.D.; Unadkat, J.D.; Slattery, J.T. Metabolism of dapsone to its hydroxylamine by CYP2E1 in vitro and in vivo. Clin. Pharmacol. Ther. 1995, 58, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.D. Dapsone: Modes of action, toxicity and possible strategies for increasing patient tolerance. Br. J. Dermatol. 1993, 129, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.D. Dapsone toxicity: Some current perspectives. Gen. Pharmacol. Vasc. Syst. 1995, 26, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Molinelli, E.; Paolinelli, M.; Campanati, A.; Brisigotti, V.; Offidani, A. Metabolic, pharmacokinetic, and toxicological issues surrounding dapsone. Expert Opin. Drug Metab. Toxicol. 2019, 15, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Wozel, G.; Schmit, J. Hypersensitivity reactions to dapsone: A systematic review. Acta Derm. Venereol. 2012, 92, 194–199. [Google Scholar] [CrossRef]
- Jouyban, A.; Rahimpour, E.; Karimzadeh, Z.; Zhao, H. Simulation of dapsone solubility data in mono- and mixed-solvents at various temperatures. J. Mol. Liq. 2022, 345, 118223. [Google Scholar] [CrossRef]
- Bergström, C.A.S.; Andersson, S.B.E.; Fagerberg, J.H.; Ragnarsson, G.; Lindahl, A. Is the full potential of the biopharmaceutics classification system reached? Eur. J. Pharm. Sci. 2014, 57, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Rauber, G.; Argenta, D.F.; Caon, T. Emerging Technologies to Target Drug Delivery to the Skin—The Role of Crystals and Carrier-Based Systems in the Case Study of Dapsone. Pharm. Res. 2020, 37, 240. [Google Scholar] [CrossRef] [PubMed]
- Mahore, J.G.; Suryawanshi, S.D.; Shirolkar, S.V.; Deshkar, S.S. Enhancement of percutaneous delivery of dapsone by microemulsion gel. J. Young Pharm. 2017, 9, 507–512. [Google Scholar] [CrossRef]
- Wozel, V.E.G. Innovative use of dapsone. Dermatol. Clin. 2010, 28, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hao, X.; Li, J.; Guan, A.; Zhou, Z.; Guo, F. New insight into improving the solubility of poorly soluble drugs by preventing the formation of their hydrogen-bonds: A case of dapsone salts with camphorsulfonic and 5-sulfosalicylic acid. CrystEngComm 2021, 23, 6191–6198. [Google Scholar] [CrossRef]
- Paredes da Rocha, N.; de Souza, A.; Nishitani Yukuyama, M.; Lopes Barreto, T.; de O. Macedo, L.; Löbenberg, R.; Lima Barros de Araújo, G.; Ishida, K.; Araci Bou-Chacra, N. Highly water-soluble dapsone nanocrystals: Towards innovative preparations for an undermined drug. Int. J. Pharm. 2023, 630, 122428. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, Y.; Xue, Y.; Zhu, P.; Zhao, H. Comprehensive insight into solubility, dissolution properties and solvation behaviour of dapsone in co-solvent solutions. J. Mol. Liq. 2021, 341, 117403. [Google Scholar] [CrossRef]
- Li, W.; Ma, Y.; Yang, Y.; Xu, S.; Shi, P.; Wu, S. Solubility measurement, correlation and mixing thermodynamics properties of dapsone in twelve mono solvents. J. Mol. Liq. 2019, 280, 175–181. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, J.; Zheng, M.; Chen, G.; Farajtabar, A.; Zhao, H. Equilibrium solubility and preferential solvation of 1,1″-sulfonylbis(4-aminobenzene) in binary aqueous solutions of n-propanol, isopropanol and 1,4-dioxane. J. Chem. Thermodyn. 2018, 122, 102–112. [Google Scholar] [CrossRef]
- Cysewski, P.; Przybyłek, M.; Jeliński, T. Intermolecular Interactions as a Measure of Dapsone Solubility in Neat Solvents and Binary Solvent Mixtures. Materials 2023, 16, 6336. [Google Scholar] [CrossRef]
- Martínez, F.; Jouyban, A.; Acree, W.E. Pharmaceuticals solubility is still nowadays widely studied everywhere. Pharm. Sci. 2017, 23, 1–2. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Kalam, M.A.; Alshamsan, A.; Alkholief, M.; Alsarra, I.A.; Ali, R.; Haq, N.; Anwer, M.K.; Shakeel, F. Solubility Measurement and Various Solubility Parameters of Glipizide in Different Neat Solvents. ACS Omega 2020, 5, 1708–1716. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, Y.; Xia, M.; Dai, W.; Zhu, B.; Mei, X. Improving the dissolution behaviors and bioavailability of abiraterone acetate via multicomponent crystal forms. Int. J. Pharm. 2022, 614, 121460. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.K.; Kumar, R.; Vipin Kumar, V.K.; Kumar, A.; Kumar Gaurav, G.; Naresh Gupta, K. A critical review on thermodynamic and hydrodynamic modeling and simulation of liquid antisolvent crystallization of pharmaceutical compounds. J. Mol. Liq. 2022, 362, 119663. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kim, C.-M.; Jo, A.-N.; Kim, J.-E. Studies on Preformulation and Formulation of JIN-001 Liquisolid Tablet with Enhanced Solubility. Pharmaceuticals 2022, 15, 412. [Google Scholar] [CrossRef] [PubMed]
- Anwer, M.K.; Muqtader, M.; Iqbal, M.; Ali, R.; Almutairy, B.K.; Alshetaili, A.; Alshahrani, S.M.; Aldawsari, M.F.; Alalaiwe, A.; Shakeel, F. Estimating the Solubility, Solution Thermodynamics, and Molecular Interaction of Aliskiren Hemifumarate in Alkylimidazolium Based Ionic Liquids. Molecules 2019, 24, 2807. [Google Scholar] [CrossRef] [PubMed]
- Constable, D.J.C.; Jimenez-Gonzalez, C.; Henderson, R.K. Perspective on solvent use in the pharmaceutical industry. Org. Process Res. Dev. 2007, 11, 133–137. [Google Scholar] [CrossRef]
- Anastas, P.T.; Williamson, T.C. Green Chemistry: An Overview. ACS Symp. Ser. 1996, 626, 1–17. [Google Scholar] [CrossRef]
- Becker, J.; Manske, C.; Randl, S. Green chemistry and sustainability metrics in the pharmaceutical manufacturing sector. Curr. Opin. Green Sustain. Chem. 2022, 33, 100562. [Google Scholar] [CrossRef]
- DeSimone, J.M. Practical approaches to green solvents. Science 2002, 297, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Jessop, P.G. Searching for green solvents. Green Chem. 2011, 13, 1391–1398. [Google Scholar] [CrossRef]
- Cvjetko Bubalo, M.; Vidović, S.; Radojčić Redovniković, I.; Jokić, S. Green solvents for green technologies. J. Chem. Technol. Biotechnol. 2015, 90, 1631–1639. [Google Scholar] [CrossRef]
- Häckl, K.; Kunz, W. Some aspects of green solvents. Comptes Rendus Chim. 2018, 21, 572–580. [Google Scholar] [CrossRef]
- Abbott, A.P.; Ahmed, E.I.; Harris, R.C.; Ryder, K.S. Evaluating water miscible deep eutectic solvents (DESs) and ionic liquids as potential lubricants. Green Chem. 2014, 16, 4156–4161. [Google Scholar] [CrossRef]
- Omar, K.A.; Sadeghi, R. Physicochemical properties of deep eutectic solvents: A review. J. Mol. Liq. 2022, 360, 119524. [Google Scholar] [CrossRef]
- Paiva, A.; Craveiro, R.; Aroso, I.; Martins, M.; Reis, R.L.; Duarte, A.R.C. Natural Deep Eutectic Solvents—Solvents for the 21st Century. ACS Sustain. Chem. Eng. 2014, 2, 1063–1071. [Google Scholar] [CrossRef]
- Zhang, Q.; De Oliveira Vigier, K.; Royer, S.; Jérôme, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef]
- Choi, Y.H.; van Spronsen, J.; Dai, Y.; Verberne, M.; Hollmann, F.; Arends, I.W.C.E.; Witkamp, G.-J.; Verpoorte, R. Are natural deep eutectic solvents the missing link in understanding cellular metabolism and physiology? Plant Physiol. 2011, 156, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural deep eutectic solvents as new potential media for green technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef]
- Espino, M.; de los Ángeles Fernández, M.; Gomez, F.J.V.; Silva, M.F. Natural designer solvents for greening analytical chemistry. TrAC Trends Anal. Chem. 2016, 76, 126–136. [Google Scholar] [CrossRef]
- Xu, G.; Shi, M.; Zhang, P.; Tu, Z.; Hu, X.; Zhang, X.; Wu, Y. Tuning the composition of deep eutectic solvents consisting of tetrabutylammonium chloride and n-decanoic acid for adjustable separation of ethylene and ethane. Sep. Purif. Technol. 2022, 298, 121680. [Google Scholar] [CrossRef]
- Cao, Y.; Tao, X.; Jiang, S.; Gao, N.; Sun, Z. Tuning thermodynamic properties of deep eutectic solvents for achieving highly efficient photothermal sensor. J. Mol. Liq. 2020, 308, 113163. [Google Scholar] [CrossRef]
- Vieira Sanches, M.; Freitas, R.; Oliva, M.; Mero, A.; De Marchi, L.; Cuccaro, A.; Fumagalli, G.; Mezzetta, A.; Colombo Dugoni, G.; Ferro, M.; et al. Are natural deep eutectic solvents always a sustainable option? A bioassay-based study. Environ. Sci. Pollut. Res. 2023, 30, 17268–17279. [Google Scholar] [CrossRef]
- Lapeña, D.; Errazquin, D.; Lomba, L.; Lafuente, C.; Giner, B. Ecotoxicity and biodegradability of pure and aqueous mixtures of deep eutectic solvents: Glyceline, ethaline, and reline. Environ. Sci. Pollut. Res. 2021, 28, 8812–8821. [Google Scholar] [CrossRef] [PubMed]
- Macário, I.P.E.; Jesus, F.; Pereira, J.L.; Ventura, S.P.M.; Gonçalves, A.M.M.; Coutinho, J.A.P.; Gonçalves, F.J.M. Unraveling the ecotoxicity of deep eutectic solvents using the mixture toxicity theory. Chemosphere 2018, 212, 890–897. [Google Scholar] [CrossRef]
- De Morais, P.; Gonçalves, F.; Coutinho, J.A.P.; Ventura, S.P.M. Ecotoxicity of Cholinium-Based Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2015, 3, 3398–3404. [Google Scholar] [CrossRef]
- Nguyen, C.-H.; Augis, L.; Fourmentin, S.; Barratt, G.; Legrand, F.-X. Deep Eutectic Solvents for Innovative Pharmaceutical Formulations. In Deep Eutectic Solvents for Medicine, Gas Solubilization and Extraction of Natural Substances; Springer: Cham, Switzerland, 2021; pp. 41–102. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Y.; Liu, J.; Wang, W.; Yang, Q.; Yang, G. Deep eutectic solvents: Recent advances in fabrication approaches and pharmaceutical applications. Int. J. Pharm. 2022, 622, 121811. [Google Scholar] [CrossRef] [PubMed]
- Pedro, S.N.; Freire, C.S.R.; Silvestre, A.J.D.; Freire, M.G. Deep Eutectic Solvents and Pharmaceuticals. Encyclopedia 2021, 1, 942–963. [Google Scholar] [CrossRef]
- Duarte, A.R.C.; Ferreira, A.S.D.; Barreiros, S.; Cabrita, E.; Reis, R.L.; Paiva, A. A comparison between pure active pharmaceutical ingredients and therapeutic deep eutectic solvents: Solubility and permeability studies. Eur. J. Pharm. Biopharm. 2017, 114, 296–304. [Google Scholar] [CrossRef]
- Pedro, S.N.; Freire, M.G.; Freire, C.S.R.; Silvestre, A.J.D. Deep eutectic solvents comprising active pharmaceutical ingredients in the development of drug delivery systems. Expert Opin. Drug Deliv. 2019, 16, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Bazzo, G.C.; Pezzini, B.R.; Stulzer, H.K. Eutectic mixtures as an approach to enhance solubility, dissolution rate and oral bioavailability of poorly water-soluble drugs. Int. J. Pharm. 2020, 588, 119741. [Google Scholar] [CrossRef] [PubMed]
- Jeliński, T.; Przybyłek, M.; Cysewski, P. Solubility advantage of sulfanilamide and sulfacetamide in natural deep eutectic systems: Experimental and theoretical investigations. Drug Dev. Ind. Pharm. 2019, 45, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Jeliński, T.; Przybyłek, M.; Cysewski, P. Natural Deep Eutectic Solvents as Agents for Improving Solubility, Stability and Delivery of Curcumin. Pharm. Res. 2019, 36, 116. [Google Scholar] [CrossRef] [PubMed]
- Jeliński, T.; Cysewski, P. Quantification of Caffeine Interactions in Choline Chloride Natural Deep Eutectic Solvents: Solubility Measurements and COSMO-RS-DARE Interpretation. Int. J. Mol. Sci. 2022, 23, 7832. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P.; Jeliński, T.; Przybyłek, M. Intermolecular Interactions of Edaravone in Aqueous Solutions of Ethaline and Glyceline Inferred from Experiments and Quantum Chemistry Computations. Molecules 2023, 28, 629. [Google Scholar] [CrossRef]
- Panapitiya, G.; Girard, M.; Hollas, A.; Sepulveda, J.; Murugesan, V.; Wang, W.; Saldanha, E. Evaluation of Deep Learning Architectures for Aqueous Solubility Prediction. ACS Omega 2022, 7, 15695–15710. [Google Scholar] [CrossRef]
- Lee, S.; Lee, M.; Gyak, K.-W.; Kim, S.D.; Kim, M.-J.; Min, K. Novel Solubility Prediction Models: Molecular Fingerprints and Physicochemical Features vs Graph Convolutional Neural Networks. ACS Omega 2022, 7, 12268–12277. [Google Scholar] [CrossRef] [PubMed]
- Hammond, O.S.; Bowron, D.T.; Edler, K.J.; Hammond, S.; Edler, K.J.; Bowron, D.T. The Effect of Water upon Deep Eutectic Solvent Nanostructure: An Unusual Transition from Ionic Mixture to Aqueous Solution. Angew. Chem. Int. Ed. 2017, 56, 9782–9785. [Google Scholar] [CrossRef]
- Jeliński, T.; Przybyłek, M.; Różalski, R.; Cysewski, P. Solubility of dapsone in deep eutectic solvents: Experimental analysis, molecular insights and machine learning predictions. Polym. Med. 2024. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Przybyłek, M. Application of COSMO-RS-DARE as a Tool for Testing Consistency of Solubility Data: Case of Coumarin in Neat Alcohols. Molecules 2022, 27, 5274. [Google Scholar] [CrossRef]
- Przybyłek, M.; Kowalska, A.; Tymorek, N.; Dziaman, T.; Cysewski, P. Thermodynamic characteristics of phenacetin in solid state and saturated solutions in several neat and binary solvents. Molecules 2021, 26, 4078. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P.; Przybyłek, M.; Rozalski, R. Experimental and Theoretical Screening for Green Solvents Improving Sulfamethizole Solubility. Materials 2021, 14, 5915. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Eckert, F. COSMO-RS: A novel and efficient method for the a priori prediction of thermophysical data of liquids. Fluid Phase Equilib. 2000, 172, 43–72. [Google Scholar] [CrossRef]
- Klamt, A. COSMO-RS from Quantum Chemistry to Fluid Phase Thermodynamics and Drug Design; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Jeliński, T.; Kubsik, M.; Cysewski, P. Application of the Solute–Solvent Intermolecular Interactions as Indicator of Caffeine Solubility in Aqueous Binary Aprotic and Proton Acceptor Solvents: Measurements and Quantum Chemistry Computations. Materials 2022, 15, 2472. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P. Intermolecular interaction as a direct measure of water solubility advantage of meloxicam cocrystalized with carboxylic acids. J. Mol. Model. 2018, 24, 112. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T. Optimization, thermodynamic characteristics and solubility predictions of natural deep eutectic solvents used for sulfonamide dissolution. Int. J. Pharm. 2019, 570, 118682. [Google Scholar] [CrossRef]
- Dassault Systèmes. COSMOtherm, Version 24.0.0, Dassault Systèmes; Biovia: San Diego, CA, USA, 2024.
- Dassault Systèmes. COSMOconf, Version 24.0.0, Dassault Systèmes; Biovia: San Diego, CA, USA, 2024.
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data mining toolbox in python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cysewski, P.; Jeliński, T.; Przybyłek, M. Experimental and Theoretical Insights into the Intermolecular Interactions in Saturated Systems of Dapsone in Conventional and Deep Eutectic Solvents. Molecules 2024, 29, 1743. https://doi.org/10.3390/molecules29081743
Cysewski P, Jeliński T, Przybyłek M. Experimental and Theoretical Insights into the Intermolecular Interactions in Saturated Systems of Dapsone in Conventional and Deep Eutectic Solvents. Molecules. 2024; 29(8):1743. https://doi.org/10.3390/molecules29081743
Chicago/Turabian StyleCysewski, Piotr, Tomasz Jeliński, and Maciej Przybyłek. 2024. "Experimental and Theoretical Insights into the Intermolecular Interactions in Saturated Systems of Dapsone in Conventional and Deep Eutectic Solvents" Molecules 29, no. 8: 1743. https://doi.org/10.3390/molecules29081743
APA StyleCysewski, P., Jeliński, T., & Przybyłek, M. (2024). Experimental and Theoretical Insights into the Intermolecular Interactions in Saturated Systems of Dapsone in Conventional and Deep Eutectic Solvents. Molecules, 29(8), 1743. https://doi.org/10.3390/molecules29081743