
Carbon Dots Anchoring Single-Atom Pt on C3N4 Boosting Photocatalytic Hydrogen Evolution

Abstract
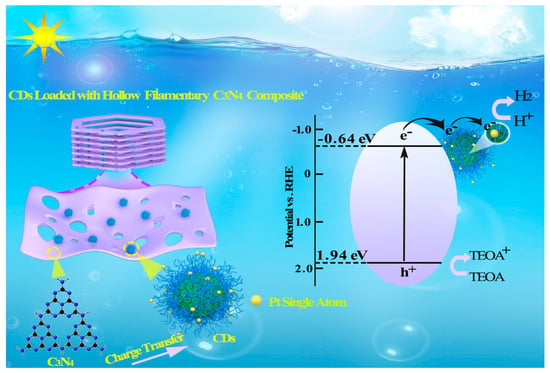
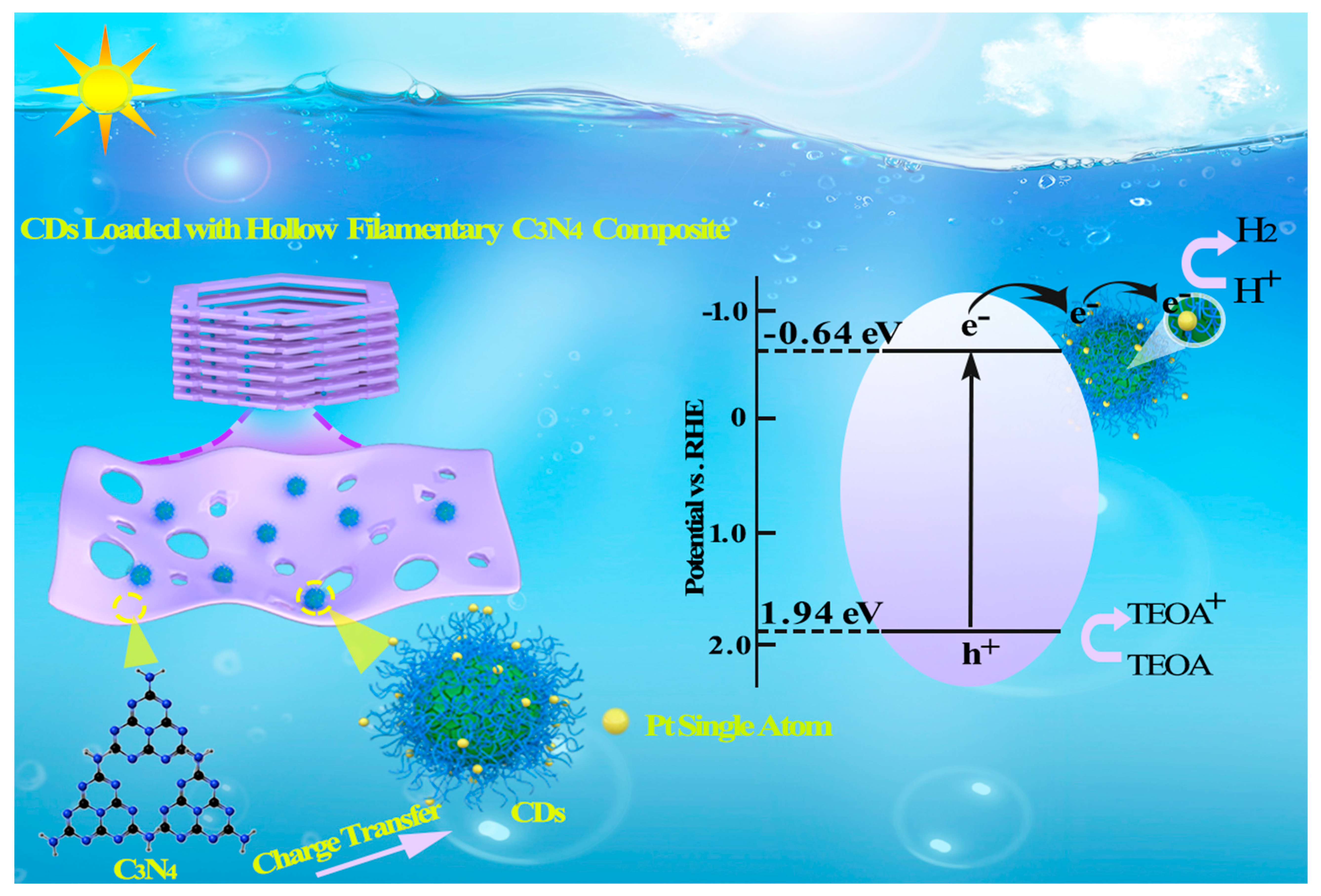
:
1. Introduction
2. Results and Discussion
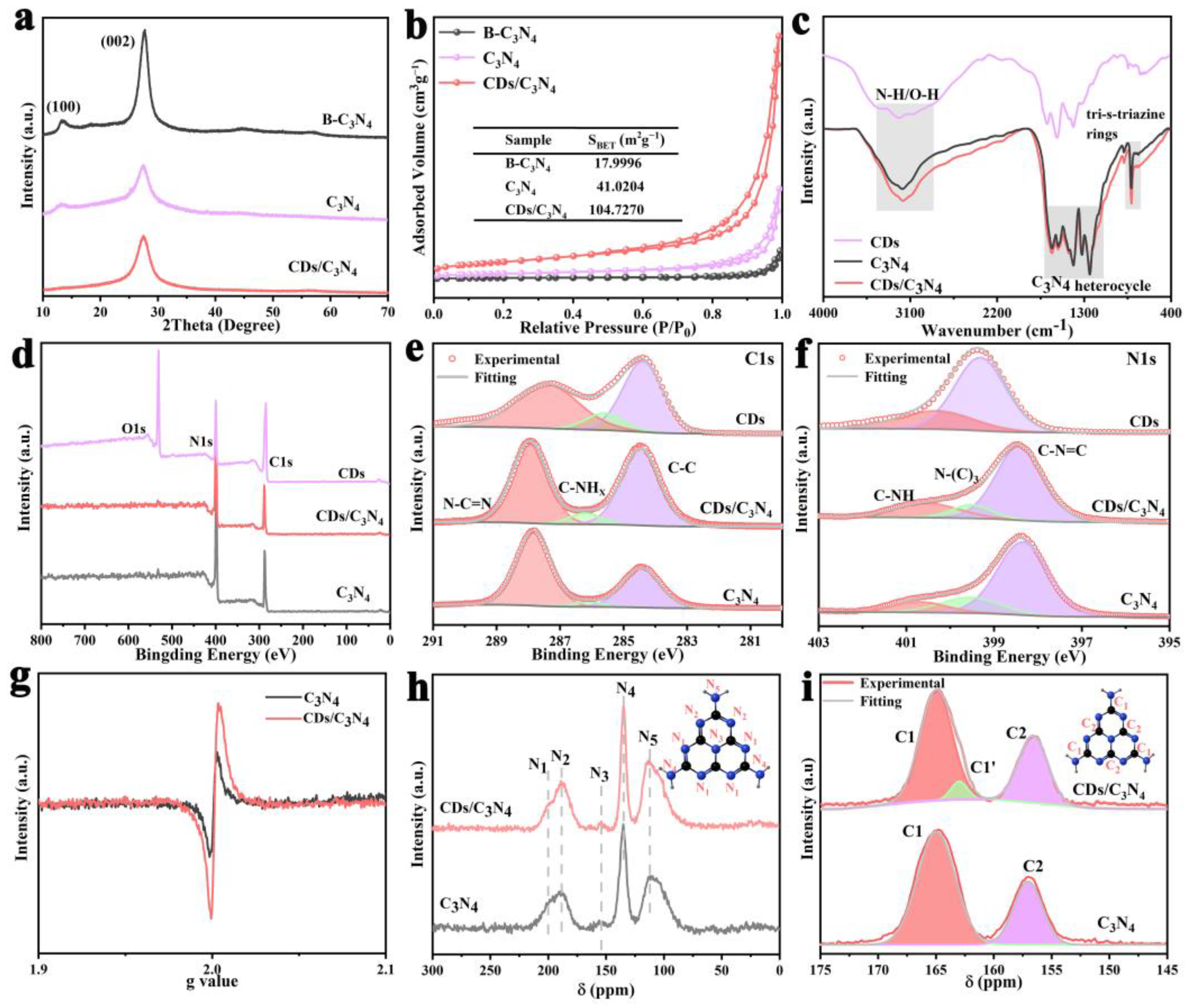
2.1. Material Preparation and Characterizations
2.2. Band Gap and Photogenerated Charge Transfer
2.3. Anchoring Effect of CDs on Pt Cocatalyst
2.4. Enhanced Photocatalytic Performance of Pt-CDs/C3N4
3. Experimental
3.1. Materials
3.2. Synthesis of Carbon Dots
3.3. Synthesis of Precursor
3.4. Synthesis of Hollow Porous CDs/C3N4
3.5. Synthesis of Pt-C3N4 and Pt-CDs/C3N4
3.6. Synthesis of C3N4
3.7. Synthesis of B-C3N4
3.8. Characterization of Sample
3.9. Photocatalytic H2 Production Experiments
3.10. Photoelectrochemical Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, H.; Sun, H.; Shi, Y.; Wang, J.; Bian, A.; Hu, Y.; Guo, F.; Shi, W.; Du, X.; Kang, Z. Cooperation of carbon doping and carbon loading boosts photocatalytic activity by the optimum photo-induced electron trapping and interfacial charge transfer. Chem. Eng. J. 2023, 472, 144654. [Google Scholar] [CrossRef]
- Gao, G.; Niu, X.; Xu, B.; Wang, X.L.; Yao, Y.-F. Shape and size effects on photocatalytic hydrogen production via Pd/C3N4 photocatalysts under visible light. Catal. Sci. Technol. 2020, 10, 5438–5442. [Google Scholar] [CrossRef]
- Zhai, B.; Li, H.; Gao, G.; Wang, Y.; Niu, P.; Wang, S.; Li, L. A Crystalline Carbon Nitride Based Near-Infrared Active Photocatalyst. Adv. Funct. Mater. 2022, 32, 7375. [Google Scholar] [CrossRef]
- Lu, X.; Xu, K.; Chen, P.; Jia, K.; Liu, S.; Wu, C. Facile one step method realizing scalable production of g-C3N4 nanosheets and study of their photocatalytic H2 evolution activity. J. Mater. Chem. A 2014, 2, 18924–18928. [Google Scholar] [CrossRef]
- Zeng, W.; Dong, Y.; Ye, X.; Guan, X.; Zhang, T.; Guo, L. Ultrathin porous carbon nitride with molecular structure regulation for excellent photocatalytic water splitting. Chem. Eng. J. 2023, 468, 143604. [Google Scholar] [CrossRef]
- Zhang, M.; Wen, J.; Zhang, S.; Zhai, Y. Tremella-like porous carbon nitride co-doped with oxygen and carbon towards efficient visible-light-driven purification of wastewater. Sep. Purif. Technol. 2021, 257, 117984. [Google Scholar] [CrossRef]
- Bao, H.; Wang, L.; Li, G.; Zhou, L.; Xu, Y.; Liu, Z.; Wu, M. Carrier engineering of carbon nitride boosts visible-light photocatalytic hydrogen evolution. Carbon 2021, 179, 80–88. [Google Scholar] [CrossRef]
- Xiao, X.; Gao, Y.; Zhang, L.; Zhang, J.; Zhang, Q.; Li, Q.; Bao, H.; Zhou, J.; Miao, S.; Chen, N.; et al. A Promoted Charge Separation/Transfer System from Cu Single Atoms and C3N4 Layers for Efficient Photocatalysis. Adv. Mater. 2020, 32, 2003082. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Zeng, Y.; Ding, M. Ordered porous nitrogen-vacancy carbon nitride for efficient visible-light hydrogen evolution. J. Colloid Interface Sci. 2023, 642, 53–60. [Google Scholar] [CrossRef]
- Xiao, Y.; Guo, S.; Tian, G.; Jiang, B.; Ren, Z.; Tian, C.; Li, W.; Fu, H. Synergetic enhancement of surface reactions and charge separation over holey C3N4/TiO2 2D heterojunctions. Sci. Bull. 2021, 66, 275–283. [Google Scholar] [CrossRef]
- Yu, W.; Zhang, T.; Zhao, Z. Garland-like intercalated carbon nitride prepared by an oxalic acid-mediated assembly strategy for highly-efficient visible-light-driven photoredox catalysis. Appl. Catal. B 2020, 278, 119342. [Google Scholar] [CrossRef]
- Xiao, Y.; Tian, G.; Li, W.; Xie, Y.; Jiang, B.; Tian, C.; Zhao, D.; Fu, H. Molecule Self-Assembly Synthesis of Porous Few-Layer Carbon Nitride for Highly Efficient Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 2508–2515. [Google Scholar] [CrossRef]
- Ran, J.; Zhang, J.; Yu, J.; Jaroniec, M.; Qiao, S.-Z. Earth-abundant cocatalysts for semiconductor-based photocatalytic water splitting. Chem. Soc. Rev. 2013, 43, 7787–7812. [Google Scholar] [CrossRef]
- Hu, Y.; Qu, Y.; Zhou, Y.; Wang, Z.; Wang, H.; Yang, B.; Yu, Z.; Wu, Y. Single Pt atom-anchored C3N4: A bridging Pt–N bond boosted electron transfer for highly efficient photocatalytic H2 generation. Chem. Eng. J. 2021, 412, 128749. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, T.; Yu, W.; Si, R.; Liu, Y.; Zhao, Z. Modulating Location of Single Copper Atoms in Polymeric Carbon Nitride for Enhanced Photoredox Catalysis. ACS Catal. 2020, 10, 5715–5722. [Google Scholar] [CrossRef]
- Li, H.; Liu, R.; Lian, S.; Liu, Y.; Huang, H.; Kang, Z. Near-infrared light controlled photocatalytic activity of carbon quantum dots for highly selective oxidation reaction. Nanoscale 2013, 5, 3289–3297. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, W.; Guo, H.; Wang, Z.; Wang, L.; Wu, M. Functional Group Modulation in Carbon Quantum Dots for Accelerating Photocatalytic CO2 Reduction. ACS Appl. Mater. Interfaces 2023, 15, 33868–33877. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Han, X.; Godin, R.; Chen, J.; Zhou, W.; Jiang, C.; Thompson, J.F.; Mustafa, K.B.; Shevlin, S.A.; et al. Unique hole-accepting carbon-dots promoting selective carbon dioxide reduction nearly 100% to methanol by pure water. Nat. Commun. 2020, 11, 2531. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Ding, S.; Chen, Y.; Liu, Y.; Fang, M.; Xiao, G.; Zhu, Y. Constructing ample active sites in nitrogen-doped carbon materials for efficient electrocatalytic carbon dioxide reduction. Nano Energy 2021, 90, 106541. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Y.; Zhai, Y.; Wen, J.; Zhang, M.; Yu, J.; Lu, S. A novel P-doped and NCDs loaded g-C3N4 with enhanced charges separation for photocatalytic hydrogen evolution. Chin. Chem. Lett. 2023, 34, 107652. [Google Scholar] [CrossRef]
- Miao, X.; Yue, X.; Ji, Z.; Shen, X.; Zhou, H.; Liu, M.; Xu, K.; Zhu, J.; Zhu, G.; Kong, L.; et al. Nitrogen-doped carbon dots decorated on g-C3N4/Ag3PO4 photocatalyst with improved visible light photocatalytic activity and mechanism insight. Appl. Catal. B 2018, 227, 459–469. [Google Scholar] [CrossRef]
- Choi, Y.; Choi, Y.; Kwon, O.H.; Kim, B.S. Carbon Dots: Bottom-Up Syntheses, Properties, and Light-Harvesting Applications. Chem. Asian J. 2018, 13, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Liu, Y.; Dimitrov, S.D.; Steier, L.; Guo, S.; Li, X.; Feng, J.; Xie, F.; Fang, Y.; Sapelkin, A.; et al. Pt single-atoms supported on nitrogen-doped carbon dots for highly efficient photocatalytic hydrogen generation. J. Mater. Chem. A 2020, 8, 14690–14696. [Google Scholar] [CrossRef]
- Yu, H.; Shi, R.; Zhao, Y.; Waterhouse, G.I.N.; Wu, L.Z.; Tung, C.H.; Zhang, T. Smart Utilization of Carbon Dots in Semiconductor Photocatalysis. Adv. Mater. 2016, 28, 9454–9477. [Google Scholar] [CrossRef]
- Chen, J.; Xiao, Y.; Wang, N.; Kang, X.; Wang, D.; Wang, C.; Liu, J.; Jiang, Y.; Fu, H. Facile synthesis of a Z-scheme CeO2/C3N4 heterojunction with enhanced charge transfer for CO2 photoreduction. Sci. China Mater. 2023, 66, 3165–3175. [Google Scholar] [CrossRef]
- Li, Q.; Jiao, Y.; Tang, Y.; Zhou, J.; Wu, B.; Jiang, B.; Fu, H. Shear Stress Triggers Ultrathin-Nanosheet Carbon Nitride Assembly for Photocatalytic H2O2 Production Coupled with Selective Alcohol Oxidation. J. Am. Chem. Soc. 2023, 245, 20837–20848. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shi, Y.; Chen, Z.; Sun, X.; Yuan, H.; Guo, F.; Shi, W. Photothermal effect of carbon dots for boosted photothermal-assisted photocatalytic water/seawater splitting into hydrogen. Chem. Eng. J. 2023, 453, 139834. [Google Scholar] [CrossRef]
- An, X.; Tang, Q.; Lan, H.; Liu, H.; Yu, X.; Qu, J.; Lin, H.; Ye, J. Facilitating Molecular Activation and Proton Feeding by Dual Active Sites on Polymeric Carbon Nitride for Efficient CO2 Photoreduction. Angew. Chem.Int. Ed. 2022, 61, e202212706. [Google Scholar] [CrossRef]
- Hu, Y.; Shim, Y.; Oh, J.; Park, S.; Park, S.; Ishii, Y. Synthesis of 13C-, 15N-Labeled Graphitic Carbon Nitrides and NMR-Based Evidence of Hydrogen-Bonding Assisted Two-Dimensional Assembly. Chem. Mater. 2017, 29, 5080–5089. [Google Scholar] [CrossRef]
- Li, X.; Sergeyev, I.V.; Aussenac, F.; Masters, A.F.; Maschmeyer, T.; Hook, J.M. Dynamic Nuclear Polarization NMR Spectroscopy of Polymeric Carbon Nitride Photocatalysts: Insights into Structural Defects and Reactivity. Angew. Chem. Int. Ed. 2018, 57, 6848–6852. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Wang, Z.; Ma, Y.; Zhou, Y.; Shi, X.; Wu, Q.; Wang, X.; Shao, M.; Huang, H.; et al. Carbon nitride assisted 2D conductive metal-organic frameworks composite photocatalyst for efficient visible light-driven H2O2 production. Appl. Catal. B 2021, 289, 120035. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Feng, Y.; Zeng, Y.; Xie, Z.; Zhang, Q.; Su, Y.; Chen, P.; Liu, Y.; Yao, K.; et al. Novel ternary photocatalyst of single atom-dispersed silver and carbon quantum dots co-loaded with ultrathin g-C3N4 for broad spectrum photocatalytic degradation of naproxen. Appl. Catal. B 2018, 221, 510–520. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Cao, D.; Wang, Y.; Zhu, Y. Peroxymonosulfate enhanced visible light photocatalytic degradation bisphenol A by single-atom dispersed Ag mesoporous g-C3N4 hybrid. Appl. Catal. B 2017, 211, 79–88. [Google Scholar] [CrossRef]
- Wang, H.; Yin, S.; Eid, K.; Li, Y.; Xu, Y.; Li, X.; Xue, H.; Wang, L. Fabrication of Mesoporous Cage-Bell Pt Nano architectonics as Efficient Catalyst for Oxygen Reduction Reaction. ACS Sustain. Chem. Eng. 2018, 6, 11768–11774. [Google Scholar]
- Zaman, S.; Su, Y.Q.; Dong, C.L.; Qi, R.; Huang, L.; Qin, Y.; Huang, Y.C.; Li, F.M.; You, B.; Guo, W.; et al. Scalable Molten Salt Synthesis of Platinum Alloys Planted in Metal–Nitrogen–Graphene for Efficient Oxygen Reduction. Angew. Chem.Int. Ed. 2021, 61, e202115835. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Cao, L.; Lu, J. Effects of the morphology and heteroatom doping of CeO2 support on the hydrogenation activity of Pt single-atoms. Catal. Sci. Technol. 2021, 11, 2844–2851. [Google Scholar] [CrossRef]
- Ma, W.; Sun, J.; Yao, S.; Wang, Y.; Chen, G.; Fan, G.; Li, Y. Synergistic Interplay of Dual-Active-Sites on Metallic Ni-MOFs Loaded with Pt for Thermal-Photocatalytic Conversion of Atmospheric CO2 under Infrared Light Irradiation. Angew. Chem. Int. Ed. 2023, 62, e202313784. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Gu, B.; Tang, Q.; Cao, Q.-E.; Fang, W. Regulating the Interaction within Pd-Cu Dual Metal Sites for Selective Hydrogenation of Furfural Using Ambient H2 Pressure. ACS Sustain. Chem. Eng. 2023, 11, 12798–12808. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Zhou, L.; Chen, C.; Han, Z.; Zhang, B.; Wu, Q.; Yang, L.; Du, L.; Bu, Y.; et al. The simplest construction of single-site catalysts by the synergism of micropore trapping and nitrogen anchoring. Nat.Commun. 2019, 10, 1657. [Google Scholar] [CrossRef]
- Yan, J.; Ji, Y.; Batmunkh, M.; An, P.; Zhang, J.; Fu, Y.; Jia, B.; Li, Y.; Liu, S.; Ye, J.; et al. Breaking Platinum Nanoparticles to Single-Atomic Pt-C4 Co-catalysts for Enhanced Solar-to-Hydrogen Conversion. Angew. Chem. Int. Ed. 2020, 60, 2541–2547. [Google Scholar] [CrossRef]
- Chen, W.; Luo, X.; Slater, T.; Zhou, Y.; Ling, S.; Bao, R.; Fernandes, J.; Shen, W.Y. General synthesis of single atom electrocatalysts via a facile condensation–carbonization process. J. Mater. Chem. A 2020, 8, 25959. [Google Scholar] [CrossRef]
- Wang, N.; Mei, R.; Lin, X.; Chen, L.; Yang, T.; Liu, Q.; Chen, Z. Cascade Anchoring Strategy for Fabricating High-Loading Pt Single Atoms as Bifunctional Catalysts for Electrocatalytic Hydrogen Evolution and Oxygen Reduction Reactions. ACS Appl. Mater. Interfaces 2023, 15, 29195–29203. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Lin, Z.; Deng, J.; Liu, Y.; Zhang, L.; Wang, K.; Xu, S.; Cao, S. Construction of carbon dots modified hollow g-C3N4 spheres via in situ calcination of cyanamide and glucose for highly enhanced visible light photocatalytic hydrogen evolution. Int. J. Hydrogen Energy 2022, 47, 1568–1578. [Google Scholar] [CrossRef]
- Yuan, H.; Shi, W.; Lu, J.; Wang, J.; Shi, Y.; Guo, F.; Kang, Z. Dual-channels separated mechanism of photo-generated charges over semiconductor photocatalyst for hydrogen evolution: Interfacial charge transfer and transport dynamics insight. Chem. Eng. J. 2023, 454, 140442. [Google Scholar] [CrossRef]
- Lv, S.; Ng, Y.H.; Zhu, R.; Li, S.; Wu, C.; Liu, Y.; Zhang, Y.; Jing, L.; Deng, J.; Dai, H. Phosphorus vapor assisted preparation of P-doped ultrathin hollow g-C3N4 sphere for efficient solar-to-hydrogen conversion. Appl. Catal. B 2021, 297, 120438. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Type and Contents of Elements (%) | ||
|---|---|---|---|
| Pt | |||
| Pt4+ | Pt2+ | Pt0 | |
| Pt-C3N4 | 17 | 63.8 | 19.2 |
| Pt-CDs/C3N4 | 16.2 | 83.8 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Song, J.; Kang, X.; Wang, D.; Tian, C.; Zhang, Q.; Zhao, H.; Liu, J. Carbon Dots Anchoring Single-Atom Pt on C3N4 Boosting Photocatalytic Hydrogen Evolution. Molecules 2024, 29, 1890. https://doi.org/10.3390/molecules29081890
Wang J, Song J, Kang X, Wang D, Tian C, Zhang Q, Zhao H, Liu J. Carbon Dots Anchoring Single-Atom Pt on C3N4 Boosting Photocatalytic Hydrogen Evolution. Molecules. 2024; 29(8):1890. https://doi.org/10.3390/molecules29081890
Chicago/Turabian StyleWang, Jing, Jiayu Song, Xin Kang, Dongxu Wang, Chungui Tian, Qin Zhang, Hui Zhao, and Jiancong Liu. 2024. "Carbon Dots Anchoring Single-Atom Pt on C3N4 Boosting Photocatalytic Hydrogen Evolution" Molecules 29, no. 8: 1890. https://doi.org/10.3390/molecules29081890
APA StyleWang, J., Song, J., Kang, X., Wang, D., Tian, C., Zhang, Q., Zhao, H., & Liu, J. (2024). Carbon Dots Anchoring Single-Atom Pt on C3N4 Boosting Photocatalytic Hydrogen Evolution. Molecules, 29(8), 1890. https://doi.org/10.3390/molecules29081890