

Quantum Size-Driven Spectral Variations in Pillar[n]arene Systems: A Density Functional Theory and Wave Function Assessment

Abstract
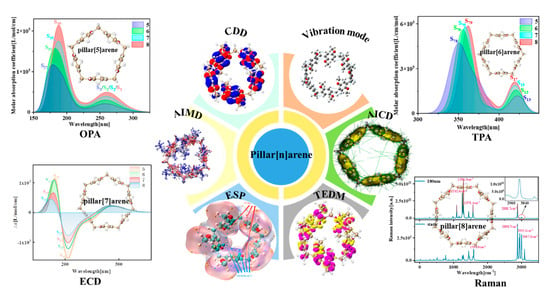
:
1. Introduction
2. Results and Discussion
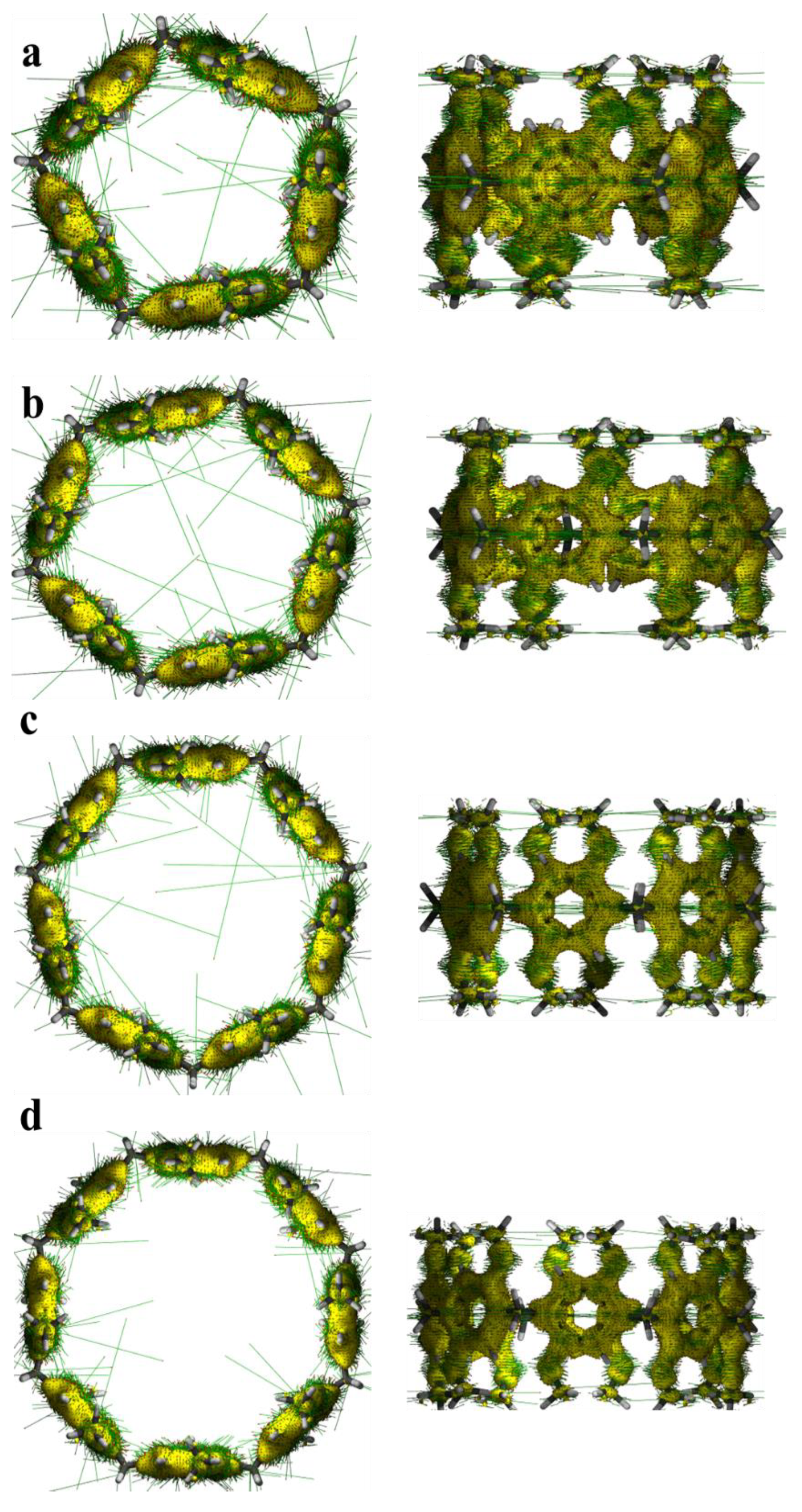
2.1. HOMO-LUMO Molecular Orbital Analysis
2.2. OPA Spectrum and TPA Spectrum Analysis
2.2.1. One-Photon Transition Characteristics
2.2.2. Two-Photon Transition Characteristics
2.3. Chiral Physical Mechanism of ECD
2.4. Raman Spectrum Analysis


2.5. ESP Analysis
2.6. Response of Pillar[n]arene to External Magnetic Field and Magnetically Induced Current Density
2.7. Molecular Dynamics Analysis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiao, T.; Qi, L.; Zhong, W.; Lin, C.; Wang, R.; Wang, L. Stimuli-responsive nanocarriers constructed from pillar[n]arene-based supra-amphiphiles. Mater. Chem. Front. 2019, 3, 1973–1993. [Google Scholar] [CrossRef]
- Ogoshi, T.; Kakuta, T.; Yamagishi, T. Applications of pillar[n]arene-based supramolecular assemblies. Angew. Chem. Int. Ed. 2019, 58, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.Y.; Yang, Y.W. Pyridine-conjugated pillar[5]arene: From molecular crystals of blue luminescence to red-emissive coordination nanocrystals. J. Am. Chem. Soc. 2021, 143, 11976–11981. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, Z.; Zhou, L.; Zhang, H.; Han, J. Recent progresses in pillar[n]arene-based photocatalysis. J. Mater. Sci. 2022, 57, 16175–16191. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Z.; Li, B.; Zhou, L.; Zhang, H.; Han, J. Hybrid Macrocyclic Polymers: Self-Assembly Containing Cucurbit[m]uril-pillar[n]arene. Polymers 2022, 14, 1777. [Google Scholar] [CrossRef]
- Khalil-Cruz, L.E.; Liu, P.; Huang, F.; Khashab, N.M. Multifunctional pillar[n]arene-based smart nanomaterials. ACS Appl. Mater. Interfaces 2021, 13, 31337–31354. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.J. The discovery of crown ethers (Noble Lecture). Angew. Chem. Int. Ed. Engl. 1988, 27, 1021–1027. [Google Scholar] [CrossRef]
- Ogoshi, T.; Kanai, S.; Fujinami, S.; Yamagishi, T.A.; Nakamoto, Y. para-Bridged symmetrical pillar[5]arenes: Their Lewis acid catalyzed synthesis and host–guest property. J. Am. Chem. Soc. 2008, 130, 5022–5023. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; You, C.C.; Zhang, H.Y. Supramolecular Chemisty-Molecular Recognition and Assembly of Synthetic Receptor; Nankai University Press: Tianjin, China, 2001. (In Chinese) [Google Scholar]
- Ogoshi, T.; Yamafuji, D.; Kotera, D.; Aoki, T.; Fujinami, S.; Yamagishi, T.A. Clickable Di-and Tetrafunctionalized Pillar[n]arenes (n = 5, 6) by Oxidation–Reduction of Pillar[n]arene Units. J. Org. Chem. 2012, 77, 11146–11152. [Google Scholar] [CrossRef]
- Lan, S.; Zhan, S.; Ding, J.; Ma, J.; Ma, D. Pillar[n]arene-based porous polymers for rapid pollutant removal from water. J. Mater. Chem. A 2017, 5, 2514–2518. [Google Scholar] [CrossRef]
- Ogoshi, T.; Yamagishi, T. Pillar[5]-and pillar[6]arene-based supramolecular assemblies built by using their cavity-size-dependent host–guest interactions. Chem. Commun. 2014, 50, 4776–4787. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Ueshima, N.; Sakakibara, F.; Yamagishi, T.A.; Haino, T. Conversion from pillar[5]arene to pillar[6–15]arenes by ring expansion and encapsulation of C60 by pillar[n]arenes with nanosize cavities. Org. Lett. 2014, 16, 2896–2899. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Kakuta, T.; Yamagishi, T.; Yang, Y.W.; Ogoshi, T. Molecular-scale porous materials based on pillar[n]arenes. Chem 2018, 4, 2029–2053. [Google Scholar] [CrossRef]
- Chen, J.F.; Ding, J.D.; Wei, T.B. Pillararenes: Fascinating planar chiral macrocyclic arenes. Chem. Commun. 2021, 57, 9029–9039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Z.; Xin, F.; Hao, A. Synthesis and application of pillar[n]arene. Chin. J. Org. Chem. 2012, 32, 219. [Google Scholar] [CrossRef]
- Strutt, N.L.; Zhang, H.; Schneebeli, S.T.; Stoddart, J.F. Amino-Functionalized Pillar[5]arene. Chem. Eur. J. 2014, 20, 10996–11004. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Hashizume, M.; Yamagishi, T.; Nakamoto, Y. Synthesis, conformational and host–guest properties of water-soluble pillar[5]arene. Chem. Commun. 2010, 46, 3708–3710. [Google Scholar] [CrossRef]
- Peerannawar, S.R.; Gejji, S.P. Electronic structure, molecular electrostatic potential and spectral characteristics of pillar[6]arene hosts and their complexes with n-octyltriethylammonium ions. Phys. Chem. Chem. Phys. 2012, 14, 8711–8722. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chi, X.; Yan, X.; Liu, J.; Yao, Y.; Chen, W.; Huang, F.; Hou, J.L. Per-hydroxylated pillar[6]arene: Synthesis, X-ray crystal structure, and host–guest complexation. Org. Lett. 2012, 14, 1532–1535. [Google Scholar] [CrossRef]
- Rowan, S.J.; Cantrill, S.J.; Cousins, G.R.L.; Sanders, J.K.; Stoddart, J.F. Dynamic covalent chemistry. Angew. Chem. Int. Ed. 2002, 41, 898–952. [Google Scholar] [CrossRef]
- Ogoshi, T.; Yamagishi, T.; Nakamoto, Y. Pillar-shaped macrocyclic hosts pillar[n]arenes: New key players for supramolecular chemistry. Chem. Rev. 2016, 116, 7937–8002. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Ueshima, N.; Akutsu, T.; Yamafuji, D.; Furuta, T.; Sakakibara, F.; Yamagishi, T.A. The template effect of solvents on high yield synthesis, co-cyclization of pillar[6]arenes and interconversion between pillar[5]-and pillar[6]arenes. Chem. Commun. 2014, 50, 5774–5777. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tao, H.Q.; Kou, Y.H.; Meier, H.; Fu, J.L. Synthesis of pillar[7]arene. Chin. Chem. Lett. 2012, 23, 509–511. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic optoelectronic materials: Mechanisms and applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.L.; Zhu, Y.; Long, H.; Jin, Y.; Zhang, W.; Yang, Y.W. Pillar[n]arene-based supramolecular organic frameworks with high hydrocarbon storage and selectivity. Chem. Commun. 2017, 53, 6409–6412. [Google Scholar] [CrossRef]
- Behera, H.; Yang, L.; Hou, J.-L. Pillar[n]arenes: Chemistry and their material applications. Chin. J. Chem. 2020, 38, 215–217. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef] [PubMed]
- Butler, W.L. Absorption spectroscopy in vivo theory and application. Annu. Rev. Plant Physiol. 1964, 15, 451–460. [Google Scholar] [CrossRef]
- Yu, S.S.; Zhang, X.Y.; Yuan, S.J.; Jiang, S.L.; Zhang, Q.; Chen, J.J.; Yu, H.Q. Electron Transfer Mechanism at the Interface of Multi-Heme Cytochromes and Metal Oxide. Adv. Sci. 2023, 10, 2302670. [Google Scholar] [CrossRef]
- Zabrodsky, H.; Avnir, D. Continuous symmetry measures. 4. Chirality. J. Am. Chem. Soc. 1995, 117, 462–473. [Google Scholar] [CrossRef]
- Naskar, S.; Saghatchi, A.; Mujica, V.; Herrmann, C. Common Trends of Chiral Induced Spin Selectivity and Optical Dichroism with Varying Helix Pitch: A First-Principles Study. Isr. J. Chem. 2022, 62, e202200053. [Google Scholar] [CrossRef]
- Das, R.S.; Agrawal, Y.K. Raman spectroscopy: Recent advancements, techniques and applications. Vib. Spectrosc. 2011, 57, 163–176. [Google Scholar] [CrossRef]
- Baughman, R.H.; Eckhardt, H.; Kertesz, M. Structure-property predictions for new planar forms of carbon: Layered phases containing sp 2 and sp atoms. J. Chem. Phys. 1987, 87, 6687–6699. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision, A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry—IV: A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Kjær, H.; Sauer, S.P.A. Pople style basis sets for the calculation of NMR spin–spin coupling constants: The 6-31G-J and 6-311G-J basis sets. J. Chem. Theory Comput. 2011, 7, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Locatelli, D.; Quici, S.; Roberto, D.; De Angelis, F. The unexpected similar second-order NLO response for nearly planar and largely twisted push–pull stilbazole chromophores: EFISH and theoretical TD-DFT evidence. Chem. Commun. 2005, 5405–5407. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Li, N.; Zhang, L.; Wang, J. Modulation of chiral spectral deflection by van der Waals force-induced molecular electropolarization in catenane oligomers. RSC Adv. 2023, 13, 11055–11061. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, C.; Wang, L.; Wang, J. Angle-resolved one and two-photon absorption spectrum in twisted bilayer graphene quantum dots. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 271, 120894. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Chen, X.; Wang, J.; Sun, M. Visualizations of electric and magnetic interactions in electronic circular dichroism and raman optical activity. J. Phys. Chem. A 2019, 123, 8071–8081. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Sun, M. The linear and non-linear optical absorption and asymmetrical electromagnetic interaction in chiral twisted bilayer graphene with hybrid edges. Mater. Today Phys. 2020, 14, 100222. [Google Scholar] [CrossRef]
- Lu, C.; Chen, P.; Sheng, H.; Li, C.; Wang, J. Physical mechanism on linear spectrum and nonlinear spectrum in double helical carbon nanomolecule–infinitene. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 282, 121674. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Sheng, H.; Wang, J. Physical mechanism on the linear spectrum and nonlinear spectrum in a twist bilayer graphdiyne nanodisk. Phys. Chem. Chem. Phys. 2023, 25, 20049–20065. [Google Scholar] [CrossRef] [PubMed]
- Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Anisotropy of the induced current density (ACID), a general method to quantify and visualize electronic delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]















{kind=link}
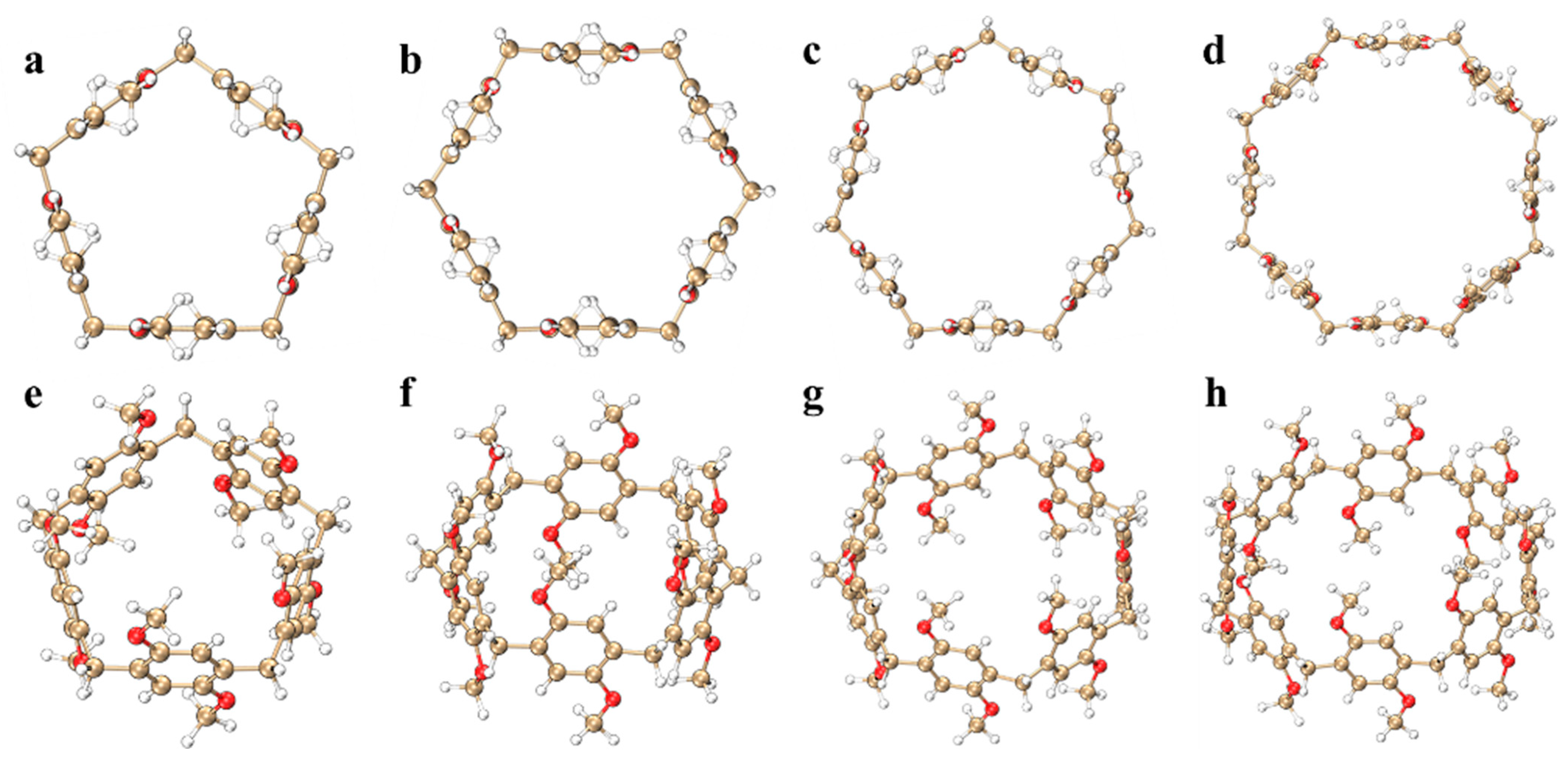{kind=link}
{kind=link}
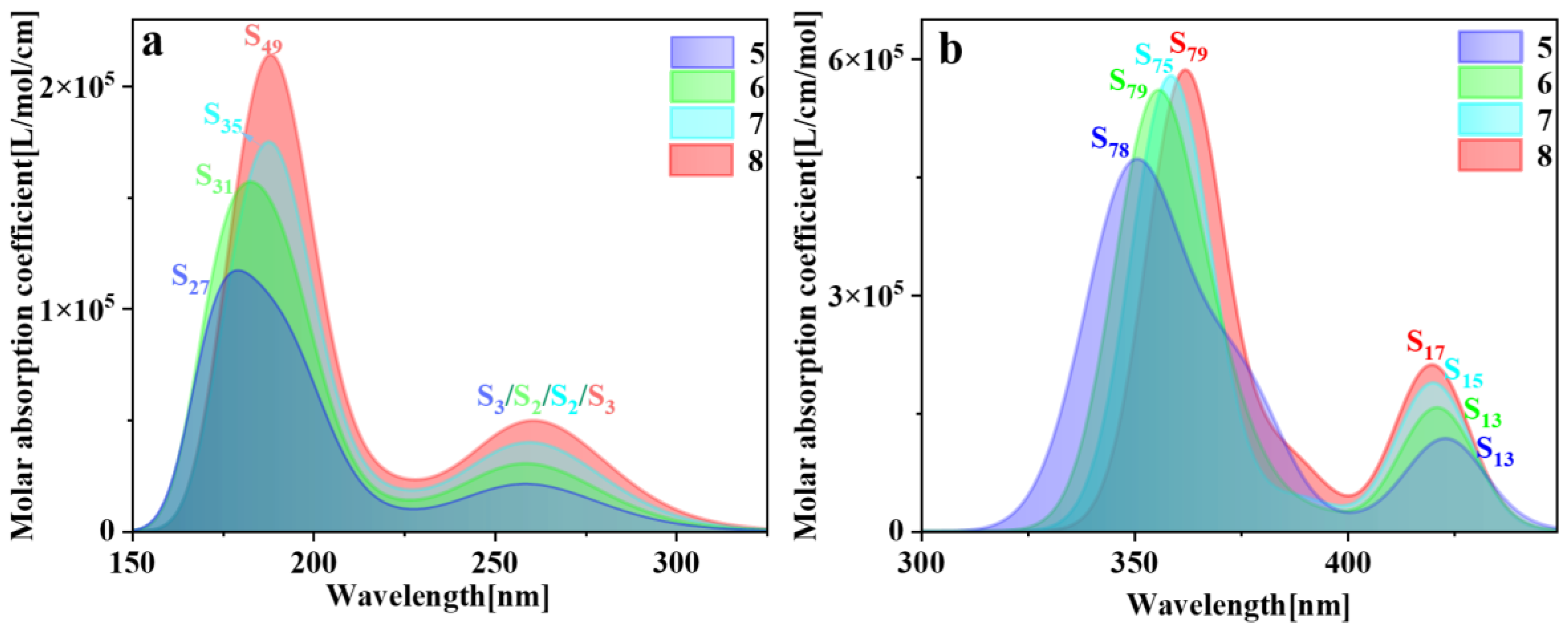{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
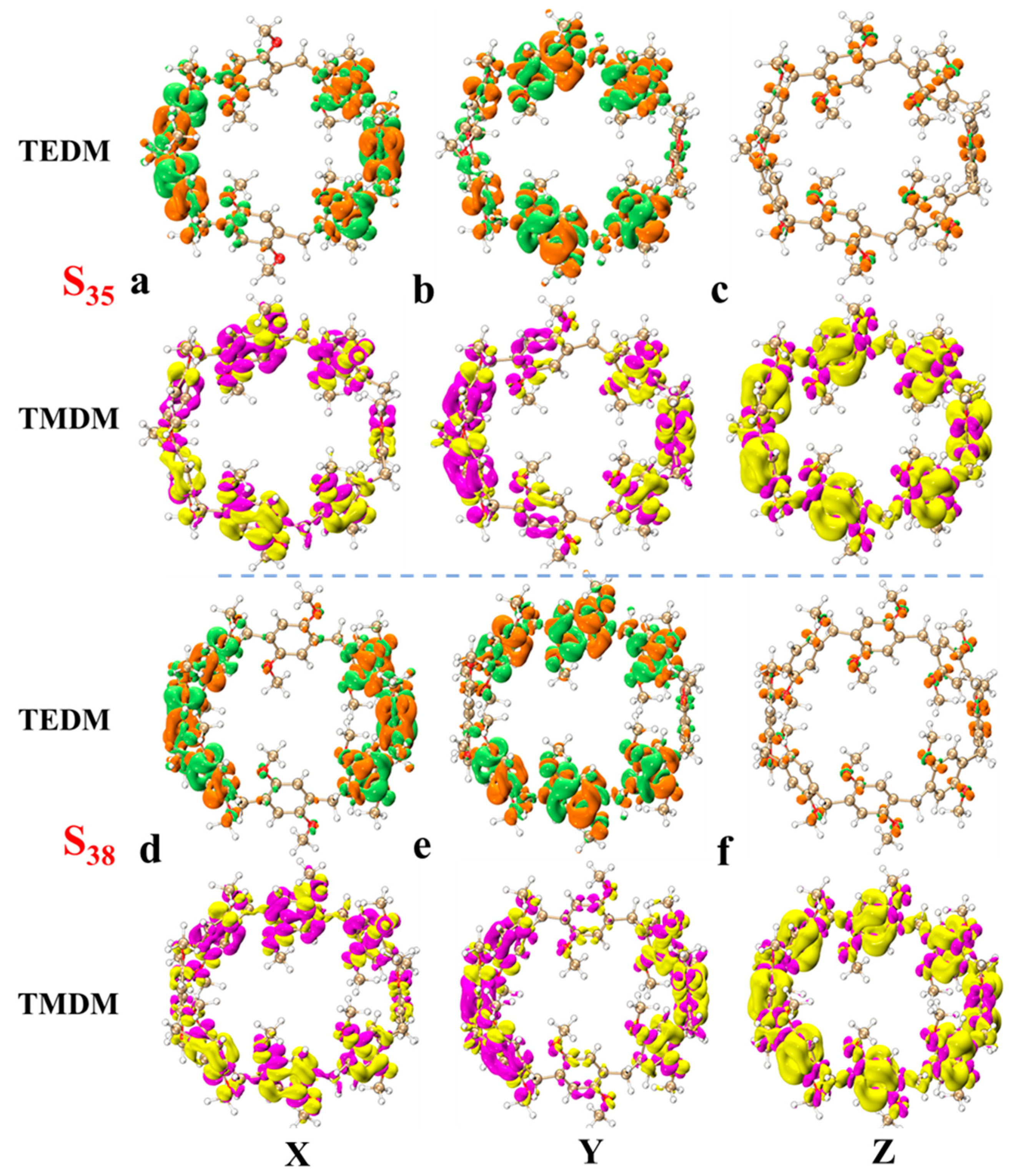{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Excited States | Oscillator Strength | Excited Energy (eV) | H (Å) | D (Å) | t (Å) | Sr |
|---|---|---|---|---|---|---|---|
| pillar[5]arene | S27 | 0.7554 | 7.187 | 4.828 | 0.089 | −1.958 | 0.80816 |
| pillar[6]arene | S31 | 0.9971 | 6.388 | 5.421 | 0.003 | −3.645 | 0.87588 |
| pillar[7]arene | S35 | 1.2394 | 6.414 | 6.254 | 0.019 | −1.629 | 0.93473 |
| pillar[8]arene | S49 | 1.5416 | 6.505 | 7.115 | 0.016 | −2.691 | 0.90027 |
| Molecule | State | Process | Integral Value (Debye) |
|---|---|---|---|
| pillar[5]arene | S78 | 4.12 × 8.11 | |
| pillar[6]arene | S79 | 2.28 × 20.38 | |
| pillar[7]arene | S75 | 0.94 × 56.23 | |
| pillar[8]arene | S79 | 0.87 × 65.99 |
| X | Y | Z | Eigenvalue | ||
|---|---|---|---|---|---|
| pillar[5]arene | TEDM | −0.0025 | −0.0010 | 2.2078 | −17.0793 |
| S27 | TMDM | 0.0020 | −0.0018 | 7.7359 | |
| pillar[6]arene | TEDM | −0.0001 | −0.0009 | 2.5240 | −27.0381 |
| S31 | TMDM | −0.0017 | −0.0013 | 10.7124 | |
| pillar[7]arene | TEDM | 0.0009 | −0.0008 | −2.8084 | −38.3265 |
| S35 | TMDM | −0.0020 | 0.0038 | −13.6471 | |
| pillar[8]arene | TEDM | 0.0149 | 0.0336 | −3.0754 | −51.0190 |
| S38 | TMDM | −0.0260 | −0.0657 | −16.5894 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, C.; Wang, T. Quantum Size-Driven Spectral Variations in Pillar[n]arene Systems: A Density Functional Theory and Wave Function Assessment. Molecules 2024, 29, 1912. https://doi.org/10.3390/molecules29091912
Yao C, Wang T. Quantum Size-Driven Spectral Variations in Pillar[n]arene Systems: A Density Functional Theory and Wave Function Assessment. Molecules. 2024; 29(9):1912. https://doi.org/10.3390/molecules29091912
Chicago/Turabian StyleYao, Cailian, and Tao Wang. 2024. "Quantum Size-Driven Spectral Variations in Pillar[n]arene Systems: A Density Functional Theory and Wave Function Assessment" Molecules 29, no. 9: 1912. https://doi.org/10.3390/molecules29091912
APA StyleYao, C., & Wang, T. (2024). Quantum Size-Driven Spectral Variations in Pillar[n]arene Systems: A Density Functional Theory and Wave Function Assessment. Molecules, 29(9), 1912. https://doi.org/10.3390/molecules29091912