

Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species


Abstract
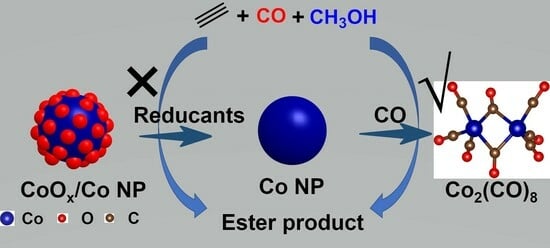
:
1. Introduction
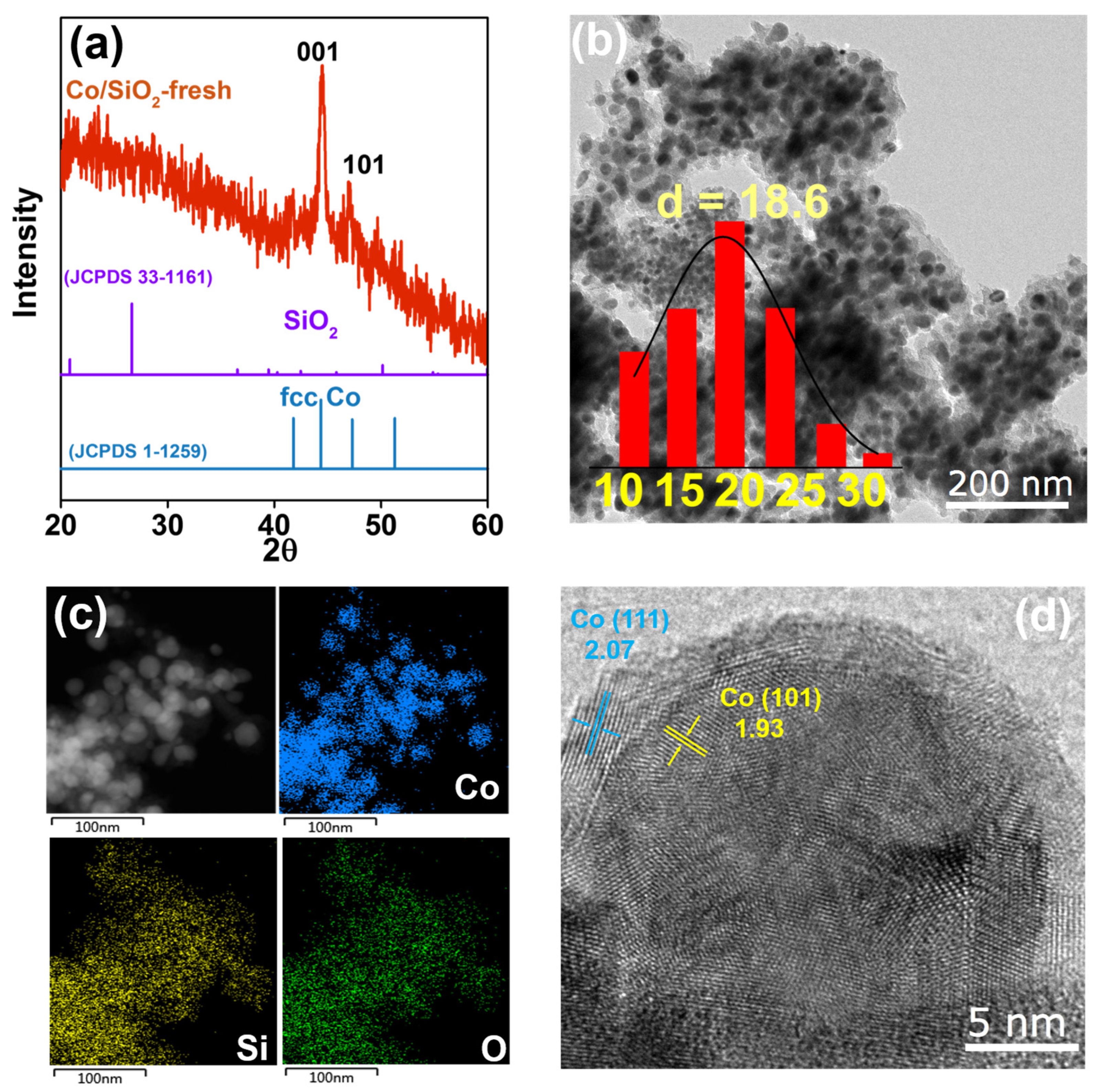
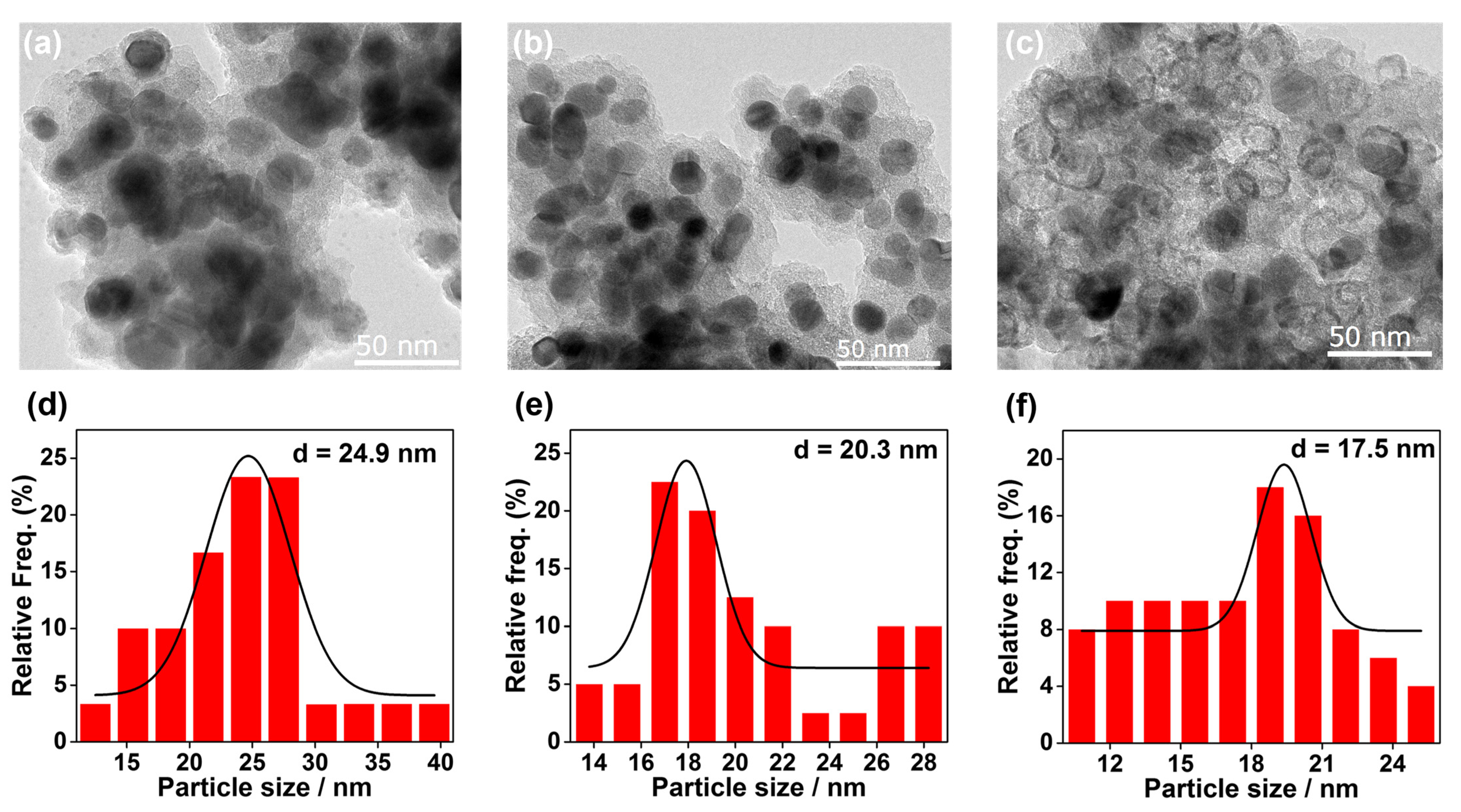
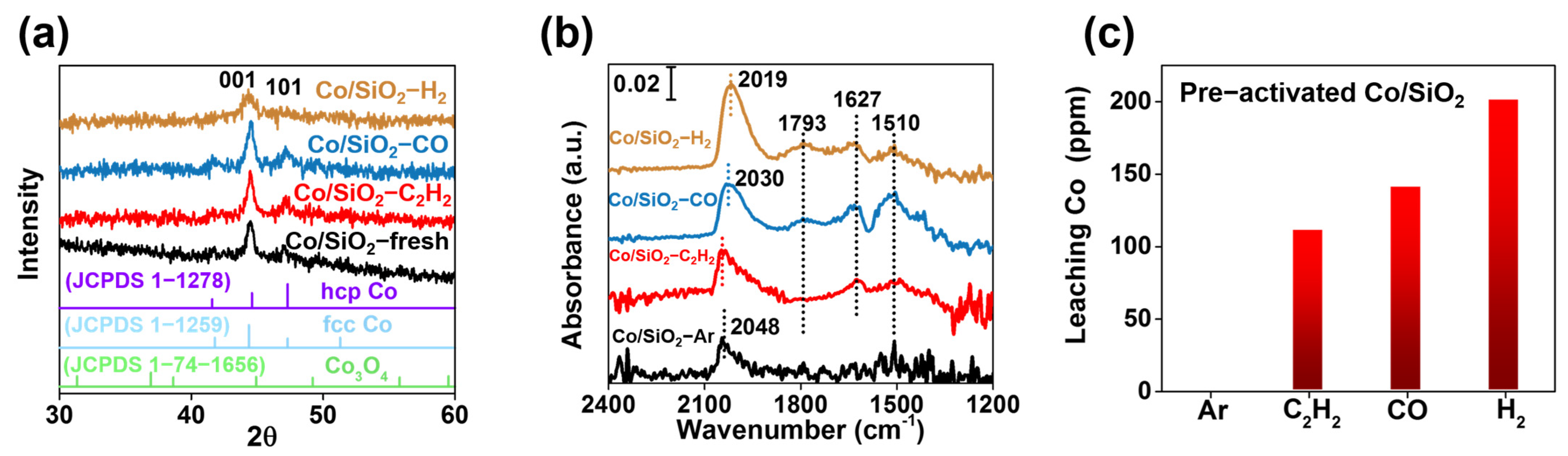
2. Results and Discussion
3. Materials and Methods
3.1. Characterization
3.2. Catalyst Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kiss, G. Palladium-catalyzed Reppe carbonylation. Chem. Rev. 2001, 101, 3435–3456. [Google Scholar] [CrossRef]
- Trotus, I.T.; Zimmermann, T.; Schueth, F. Catalytic Reactions of Acetylene: A Feedstock for the Chemical Industry Revisited. Chem. Rev. 2014, 114, 1761–1782. [Google Scholar] [CrossRef] [PubMed]
- Kalck, P.; Urrutigoïty, M. Recent improvements in the alkoxycarbonylation reaction catalyzed by transition metal complexes. Inorg. Chim. Acta 2015, 431, 110–121. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. Chemicals from Alkynes with Palladium Catalysts. Chem. Rev. 2014, 114, 1783–1826. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Wang, J.Q.; Yuan, Q.; Song, X.E.; Mu, J.L.; Wei, Y.; Yan, L.; Sun, F.F.; Feng, S.Q.; Cai, Y.T.; et al. Palladium and Ruthenium Dual-Single-Atom Sites on Porous Ionic Polymers for Acetylene Dialkoxycarbonylation: Synergetic Effects Stabilize the Active Site and Increase CO Adsorption. Angew. Chem. Int. Ed. 2023, 62, 202307570. [Google Scholar] [CrossRef] [PubMed]
- Reppe, W. New developments in the field of chemical acetylene and coal. Experientia 1949, 5, 93–110. [Google Scholar] [CrossRef]
- Bhattacharyya, S.K.; Sen, A.K. Catalytic Syntheses of Acrylic Acid and Ethyl Acrylate from Acetylene, Carbon Monoxide, and Water or Ethanol under Pressure. Ind. Eng. Chem. Process Des. Dev. 1964, 3, 169–176. [Google Scholar] [CrossRef]
- Bhattacharyya, S.K.; Nag, S.N. Catalytic synthesis of ethyl propionate from ethylene, carbon monoxide and ethanol at high pressure. J. Appl. Chem. 1962, 12, 182. [Google Scholar] [CrossRef]
- Cui, L.; Yang, X.G.; Zeng, Y.; Chen, Y.T.; Wang, G.Y. A unique nickel-base nitrogen-oxygen bidentate ligand catalyst for carbonylation of acetylene to acrylic acid. Mol. Catal. 2019, 468, 57–61. [Google Scholar] [CrossRef]
- Tang, C.M.; Zeng, Y.; Cao, P.; Yang, X.G.; Wang, G.Y. The Nickel and Copper-Catalyzed Hydroformylation of Acetylene with Carbon Monoxide to Acrylic Acid. Catal. Lett. 2009, 129, 189–193. [Google Scholar] [CrossRef]
- Lin, T.J.; Meng, X.; Shi, L. Catalytic hydrocarboxylation of acetylene to acrylic acid using Ni2O3 and cupric bromide as combined catalysts. J. Mol. Catal. A Chem. 2015, 396, 77–83. [Google Scholar] [CrossRef]
- Li, Y.K.; Yan, L.F.; Zhang, Q.F.; Yan, B.H.; Cheng, Y. A recyclable heterogeneous-homogeneous-heterogeneous NiO/AlOOH catalysis system for hydrocarboxylation of acetylene to acrylic acid. RSC Adv. 2020, 10, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.K.; Bhattach, D.P. Catalytic synthesis of n-propyl acrylate from acetylene carbon monoxide and n-propanol under pressure. J. Appl. Chem. 1966, 16, 18. [Google Scholar] [CrossRef]
- Hu, G.; Guo, D.; Shang, H.J.; Sun, Y.K.; Zeng, J.M.; Li, J.B.; Zhu, M.Y. Expanded Two-Dimensional Layered Vermiculite Supported Nickel Oxide Nanoparticles Provides High Activity for Acetylene Carbonylation to Synthesize Acrylic Acid. Catal. Lett. 2020, 150, 674–682. [Google Scholar] [CrossRef]
- Hu, G.; Guo, D.; Shang, H.J.; Sun, Y.K.; Zeng, J.M.; Li, J.B. Microwave-Assisted Rapid Preparation of Vermiculite-Loaded Nano-Nickel Oxide As a Highly Efficient Catalyst for Acetylene Carbonylation to Synthesize Acrylic Acid. Chem. Select. 2020, 5, 2940–2948. [Google Scholar] [CrossRef]
- Xie, H.; Lin, T.J.; Shi, L.; Meng, X. Acetylene carbonylation over Ni-containing catalysts: Role of surface structure and active site distribution. RSC Adv. 2016, 6, 97285–97292. [Google Scholar] [CrossRef]
- Lin, T.J.; Meng, X.; Shi, L. Ni-exchanged Y-zeolite. An efficient heterogeneous catalyst for acetylene hydrocarboxylation. Appl. Catal. A 2014, 485, 163–171. [Google Scholar] [CrossRef]
- Wu, X.F.; Fang, X.J.; Wu, L.P.; Jackstell, R.; Neumann, H.; Beller, M. Transition-Metal-Catalyzed Carbonylation Reactions of Olefins and Alkynes: A Personal Account. Acc. Chem. Res. 2014, 47, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Arnoldy, P.; Budzelaar, P.H.M. Homogeneous catalysis by cationic palladium complexes precision catalysis in the carbonylation of alkynes. J. Organomet. Chem. 1994, 475, 57–63. [Google Scholar] [CrossRef]
- Wei, X.M.; Ma, Z.W.; Mu, X.Y.; Lu, J.Z.; Hu, B. Catalyst in Acetylene Carbonylation: From Homogeneous to Heterogeneous. Prog. Chem. 2021, 33, 243–253. [Google Scholar] [CrossRef]
- Chen, X.K.; Zhu, H.J.; Wang, W.L.; Du, H.; Wang, T.; Yan, L.; Hu, X.P.; Ding, Y.J. Multifunctional Single-Site Catalysts for Alkoxycarbonylation of Terminal Alkynes. ChemSusChem 2016, 9, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.K.; Zhu, H.J.; Wang, T.; Li, C.Y.; Yan, L.; Jiang, M.; Liu, J.; Sun, X.P.; Jiang, Z.; Ding, Y.J. The 2V-P,N polymer supported palladium catalyst for methoxycarbonylation of acetylene. J. Mol. Catal. A Chem. 2016, 414, 37–46. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, L.L.; Yu, Z.A.; Zhang, S.X.; Li, L.; Ren, Y.J.; Yang, J.; Liu, X.Y.; Liu, W.; Yang, X.F.; et al. Ethylene Methoxycarbonylation over Heterogeneous Pt1/MoS2 Single-Atom Catalyst: Metal-Support Concerted Catalysis. J. Am. Chem. Soc. 2023, 146, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Feng, S.Q.; Hemberger, P.; Bodi, A.; Song, X.G.; Yuan, Q.; Mu, J.L.; Li, B.; Jiang, Z.; Ding, Y.J. Iodide-Coordinated Single-Site Pd Catalysts for Alkyne Dialkoxycarbonylation. ACS Catal. 2021, 11, 9242–9251. [Google Scholar] [CrossRef]
- Sarkar, B.R.; Chaudhari, R.V. Carbonylation of alkynes, alkenes and alcohols using metal complex catalysts. Catal. Surv. Asia 2005, 9, 193–205. [Google Scholar] [CrossRef]
- Zhang, X.H.; Shen, C.R.; Xia, C.G.; Tian, X.X.; He, L. Alkoxycarbonylation of olefins with carbon dioxide by a reusable heterobimetallic ruthenium-cobalt catalytic system. Green Chem. 2018, 20, 5533–5539. [Google Scholar] [CrossRef]
- Pesa, F.; Haase, T. The cobalt-catalyzed hydroesterification of acrylonitrile. J. Mol. Catal. 1983, 18, 237–249. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhou, H.; Deng, L.; Jia, X.; Li, Y. Molybdenum disulfide promoted co-catalyzed alkoxycarbonylation. J. Catal. 2024, 430, 115349. [Google Scholar] [CrossRef]
- Hofmann, P.; Kosswig, K.; Schaefer, W. Hydrocarboxymethylation—An attractive route from olefins to fatty-acid esters? Ind. Eng. Chem. Prod. Res. Dev. 1980, 19, 330–334. [Google Scholar] [CrossRef]
- Huang, W.H.; Jackstell, R.; Spannenberg, A.; Beller, M. An improved cobalt-catalysed alkoxycarbonylation of olefins using secondary phosphine oxide promotors. Catal. Sci. Technol. 2023, 13, 2475–2479. [Google Scholar] [CrossRef]
- Orchin, M. Hydrogenation of organic compounds with synthesis gas. Adv. Catal. 1953, 5, 385–415. [Google Scholar] [CrossRef]
- Satyanarayana, N.; Periasamy, M. Carbonylation of benzyl halides using CoCl2/NaBH4/CO/NaOH reagent system. Tetrahedron Lett. 1987, 28, 2633–2636. [Google Scholar] [CrossRef]
- Itoh, K.; Miura, M.; Nomura, M. Normal pressure double carbonylation of aryl halides using Cobalt(ii) chloride in the presence of either sodium sulfide or sodium-borohydride. Bull. Chem. Soc. Jpn. 1988, 61, 4151–4152. [Google Scholar] [CrossRef]
- Wang, L.X.; Guan, E.J.; Wang, Y.Q.; Wang, L.; Gong, Z.M.; Cui, Y.; Meng, X.J.; Gates, B.C.; Xiao, F.S. Silica accelerates the selective hydrogenation of CO2 to methanol on cobalt catalysts. Nat. Commun. 2020, 11, 14817. [Google Scholar] [CrossRef] [PubMed]
- Muniz, F.T.L.; Miranda, M.A.R.; dos Santos, C.M.; Sasaki, J.M. The Scherrer equation and the dynamical theory of X-ray diffraction. Acta Crystallogr. Sect. A Found. Adv. 2016, 72, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Kaneti, Y.V.; Zhang, J.; He, Y.B.; Wang, Z.J.; Tanaka, S.; Hossain, M.S.A.; Pan, Z.Z.; Xiang, B.; Yang, Q.H.; Yamauchi, Y. Fabrication of an MOF-derived heteroatom-doped Co/CoO/carbon hybrid with superior sodium storage performance for sodium-ion batteries. J. Mater. Chem. 2017, 5, 15356–15366. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Deng, W.P.; Wang, Y. Recent advances in understanding the key catalyst factors for Fischer-Tropsch synthesis. J. Energy Chem. 2013, 22, 27–38. [Google Scholar] [CrossRef]
- Guan, D.Q.; Zhong, J.; Xu, H.Y.; Huang, Y.C.; Hu, Z.W.; Chen, B.; Zhang, Y.; Ni, M.; Xu, X.M.; Zhou, W.; et al. A universal chemical-induced tensile strain tuning strategy to boost oxygen-evolving electrocatalysis on perovskite oxides. Appl. Phys. Res. 2022, 9, 011422. [Google Scholar] [CrossRef]
- LaGrow, A.P.; Lloyd, D.C.; Gai, P.L.; Boyes, E.D. In Situ Scanning Transmission Electron Microscopy of Ni Nanoparticle Redispersion via the Reduction of Hollow NiO. Chem. Mater. 2018, 30, 197–203. [Google Scholar] [CrossRef]
- Du, H.; Jiang, M.; Zhu, H.J.; Huang, C.D.; Zhao, Z.; Dong, W.D.; Lu, W.; Liu, T.; Zhang, Z.C.; Ding, Y.J. Constructing efficient hcp-Co active sites for Fischer-Tropsch reaction on an activated carbon supported cobalt catalyst via multistep activation processes. Fuel 2021, 292, 120244. [Google Scholar] [CrossRef]
- Parastaev, A.; Muravev, V.; Osta, E.H.; van Hoof, A.J.F.; Kimpel, T.F.; Kosinov, N.; Hensen, E.J.M. Boosting CO2 hydrogenation via size-dependent metal-support interactions in cobalt/ceria-based catalysts. Nat. Catal. 2020, 3, 526–533. [Google Scholar] [CrossRef]
- Parastaev, A.; Muravev, V.; Osta, E.H.; Kimpel, T.F.; Simons, J.F.M.; van Hoof, A.J.F.; Uslamin, E.; Zhang, L.; Struijs, J.J.C.; Burueva, D.B.; et al. Breaking structure sensitivity in CO2 hydrogenation by tuning metal-oxide interfaces in supported cobalt nanoparticles. Nat. Catal. 2022, 5, 1051–1060. [Google Scholar] [CrossRef]
- Weststrate, C.J.; van de Loosdrecht, J.; Niemantsverdriet, J.W. Spectroscopic insights into cobalt-catalyzed Fischer-Tropsch synthesis: A review of the carbon monoxide interaction with single crystalline surfaces of cobalt. J. Catal. 2016, 342, 1–16. [Google Scholar] [CrossRef]
- Zhu, H.Q.; Qin, Z.F.; Shan, W.J.; Shen, W.J.; Wang, J.G. Pd/CeO2-TiO2 catalyst for CO oxidation at low temperature:: A TPR study with H2 and CO as reducing agents. J. Catal. 2004, 225, 267–277. [Google Scholar] [CrossRef]
- Tabakova, T.; Boccuzzi, F.B.; Manzoli, M.; Andreeva, D. FTIR study of low-temperature water-gas shift reaction on gold/ceria catalyst. Appl. Catal. A 2003, 252, 385–397. [Google Scholar] [CrossRef]
- Banham, D.; Ye, S.; Pei, K.; Ozaki, J.; Kishimoto, T.; Imashiro, Y. A review of the stability and durability of non-precious metal catalysts for the oxygen reduction reaction in proton exchange membrane fuel cells. J. Power Sources 2015, 285, 334–348. [Google Scholar] [CrossRef]
- Hertrich, M.F.; Scharnagl, F.K.; Pews-Davtyan, A.; Kreyenschulte, C.R.; Lund, H.; Bartling, S.; Jackstell, R.; Beller, M. Supported Cobalt Nanoparticles for Hydroformylation Reactions. Chem. Eur. J. 2019, 25, 5534–5538. [Google Scholar] [CrossRef]
- Zhao, J.J.; He, Y.R.; Wang, F.; Zheng, W.T.; Huo, C.F.; Liu, X.; Jiao, H.J.; Yang, Y.; Li, Y.W.; Wen, X.D. Suppressing Metal Leaching in a Supported Co/SiO2 Catalyst with Effective Protectants in the Hydroformylation Reaction. ACS Catal. 2020, 10, 914–920. [Google Scholar] [CrossRef]
- Rush, L.E.; Pringle, P.G.; Harvey, J.N. Computational Kinetics of Cobalt-Catalyzed Alkene Hydroformylation. Angew. Chem. Int. Ed. 2014, 53, 8672–8676. [Google Scholar] [CrossRef]
- Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Hood, D.M.; Johnson, R.A.; Carpenter, A.E.; Younker, J.M.; Vinyard, D.J.; Stanley, G.G. Highly active cationic cobalt(II) hydroformylation catalysts. Science 2020, 367, 542–549. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Product Rate (gproduct gcat−1 h−1) | Yield % | |||
|---|---|---|---|---|---|---|
| MA | MP | DMS | Others | |||
| 1 | a 41.7% Co/SiO2 | 0 | 0 | 0 | 0 | 0 |
| 2 | a 31.6% Co/SiO2 | 0 | 0 | 0 | 0 | 0 |
| 3 | a 25.6% Co/SiO2 | 0 | 0 | 0 | 0 | 0 |
| 4 | b 41.7% Co/SiO2 | 0 | 4.42 | 0.58 | 0.49 | 64.1 |
| 5 | b 31.6% Co/SiO2 | 0.43 | 4.0 | 0.56 | 0.39 | 58.6 |
| 6 | b 25.6% Co/SiO2 | 0.16 | 0.23 | 0 | 0 | ≈0 |
| 7 | b 5% Co/WOx | 0 | 0 | 0 | 0 | 0 |
| 8 | b 5% Co/HAP | 0 | 0 | 0 | 0 | 0 |
| 9 | b 5% Co/TiO2 | 0 | 0 | 0 | 0 | 0 |
| 10 | b Co/S-1 | 0 | 0 | 0 | 0 | 0 |
| 11 | b Co powder | 0 | 0 | 0 | 0 | 0 |
| Entry | Pre-Activation | Product Rate (gproduct gcat−1 h−1) | Reaction Rate (molMA+MP+DMS molCo h−1) | ||
|---|---|---|---|---|---|
| MA | MP | DMS | |||
| 1 | 1 MPa Ar | 0 | 0 | 0 | 0 |
| 2 | 0.05 MPa C2H2 | 1.51 | 0 | 0.42 | 0.31 |
| 3 | 4 MPa CO | 2.91 | 0.03 | 0.54 | 0.56 |
| 4 | 1 MPa H2 | 4.23 | 0.15 | 0.91 | 0.85 |
| 5 | 1 MPa H2 + 4 MPa CO | 3.4 | 0.16 | 1.25 | 0.77 |
| 6 | 0.01 MPa NH3 | 0 | 0 | 0 | 0 |
| Catalyst | Product Rate (gproduct gcat−1 h−1) | ||
|---|---|---|---|
| MA | MP | DMS | |
| a Filtrate | 3.6 | 0.16 | 0.35 |
| a Residual solid | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, A.; Cao, H.; Zhang, L.; Wang, A. Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species. Molecules 2024, 29, 1987. https://doi.org/10.3390/molecules29091987
Wang A, Cao H, Zhang L, Wang A. Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species. Molecules. 2024; 29(9):1987. https://doi.org/10.3390/molecules29091987
Chicago/Turabian StyleWang, An, Hongchen Cao, Leilei Zhang, and Aiqin Wang. 2024. "Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species" Molecules 29, no. 9: 1987. https://doi.org/10.3390/molecules29091987
APA StyleWang, A., Cao, H., Zhang, L., & Wang, A. (2024). Co/SiO2 Catalyst for Methoxycarbonylation of Acetylene: On Catalytic Performance and Active Species. Molecules, 29(9), 1987. https://doi.org/10.3390/molecules29091987