A Short Review on Cardiotonic Steroids and Their Aminoguanidine Analogues

Abstract
:| 1. Introduction | 3.2.1. Structure-activity relationships |
| 2. Mechanism of inotropic activity | 3.2.2. Steroidal framework |
| 2.1. Na+,K+-ATPase | 3.2.3. Side chain at C-17 |
| 2.2. Isoenzymes | 3.2.4. Sugar |
| 3. Compounds displaying inotropic digitalis-like activity | 3.2.5. Other substituents at position C-3 |
| 3.1. Natural digitalis-like compounds | 3.2.6. Pregnane derivatives |
| 3.1.1. Digitalis-like glycosides in vegetal species | 3.3. Endogenous digitalis-like factors |
| 3.1.2. Digitalis-like glycosides in animal species | 4. Digitalis analogues bearing aminoguanidine moieties |
| 3.1.3. Natural digitalis-like glycosides recently isolated | 4.1. Aminoguanidine moiety in drugs |
| 3.2. Semisynthetic and synthetic digitalis-like compounds | 4.2. Aminoguanidines in digitalis compounds |
1. Introduction
2. Mechanism of Inotropic Activity
2.1. Na+,K+-ATPase
2.2. Isoenzymes
3. Compounds Displaying Inotropic, Digitalis-like Activity
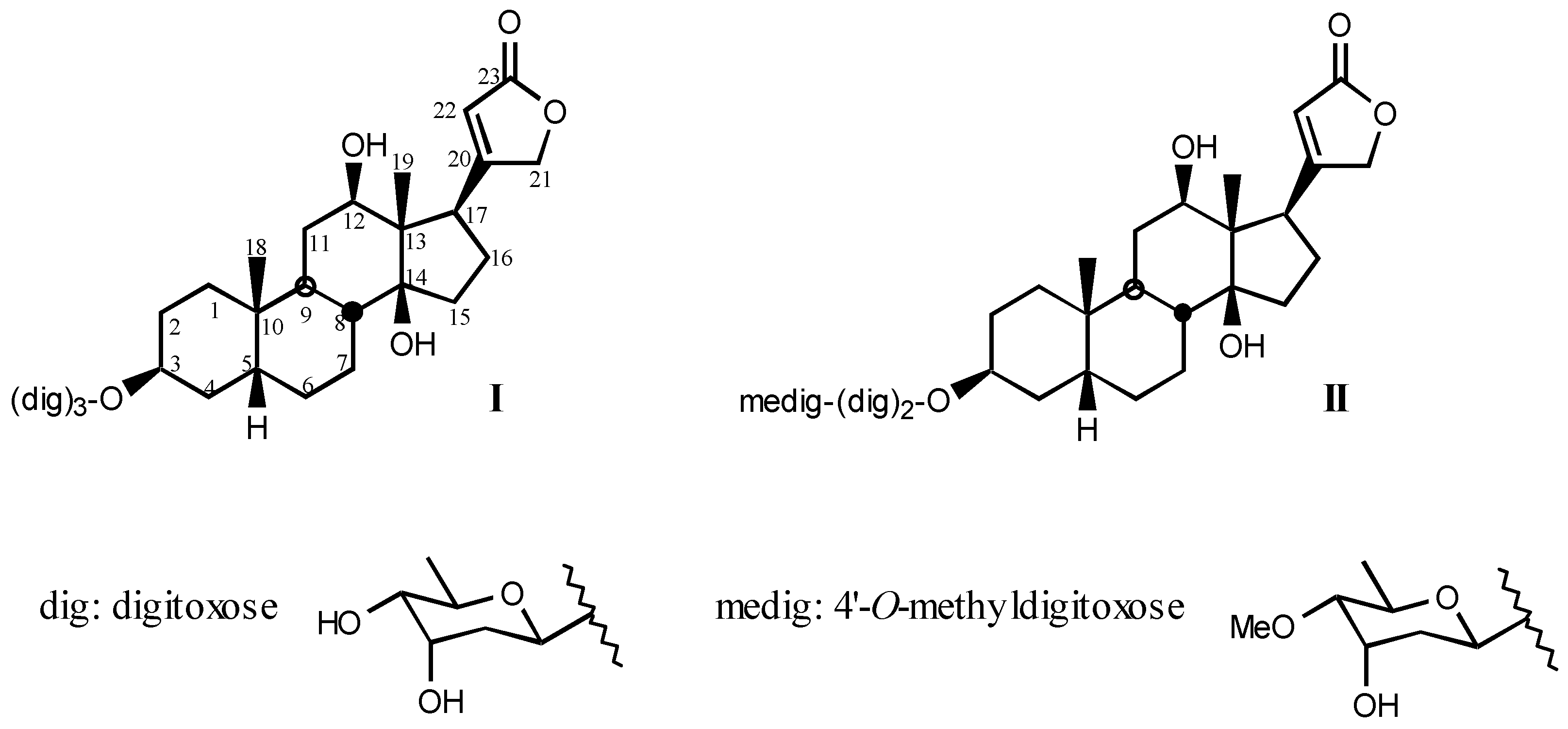
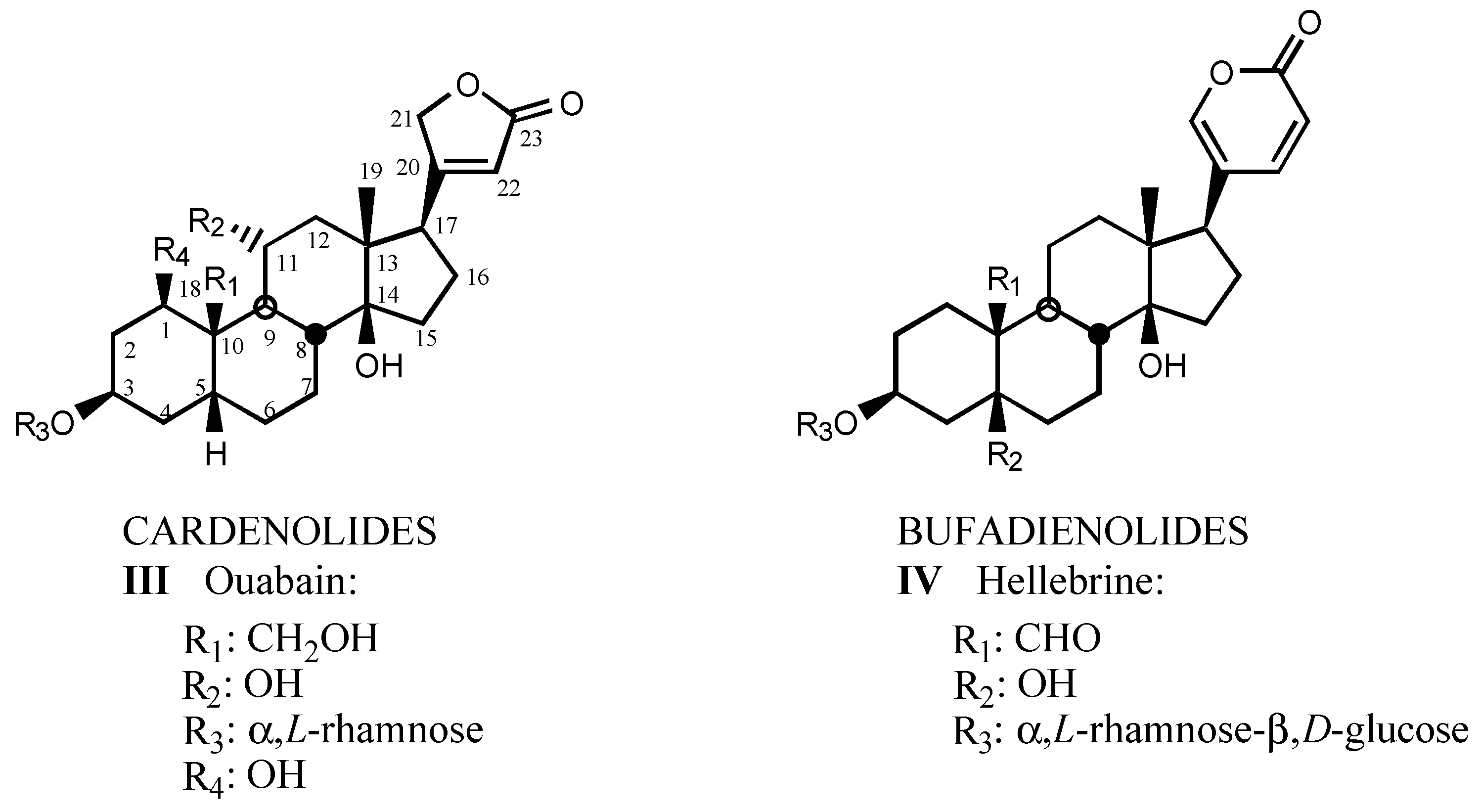
3.1. Natural digitalis-like compounds

3.1.1. Digitalis-like glycosides in vegetal species
3.1.2. Digitalis-like glycosides in animal species
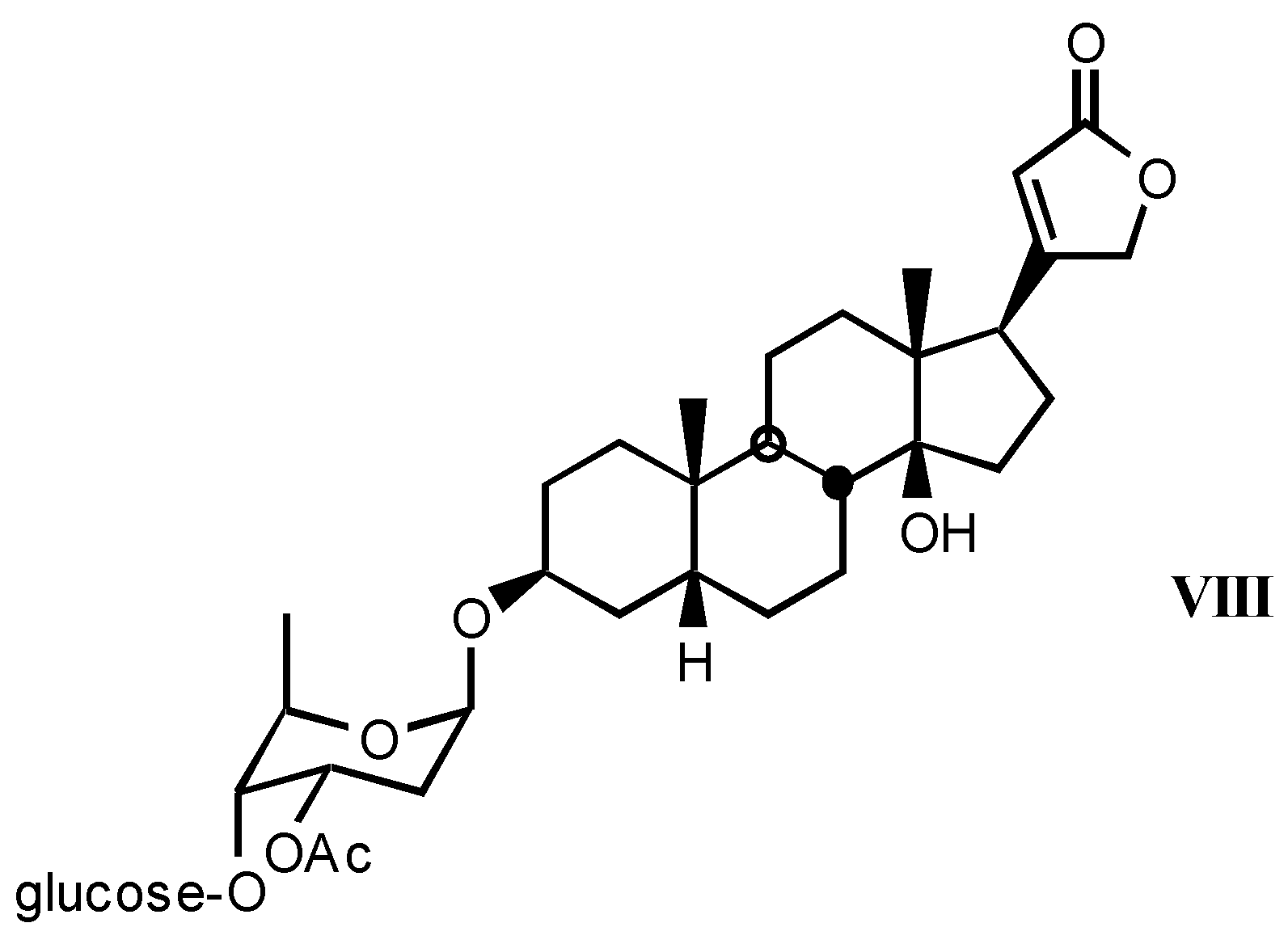
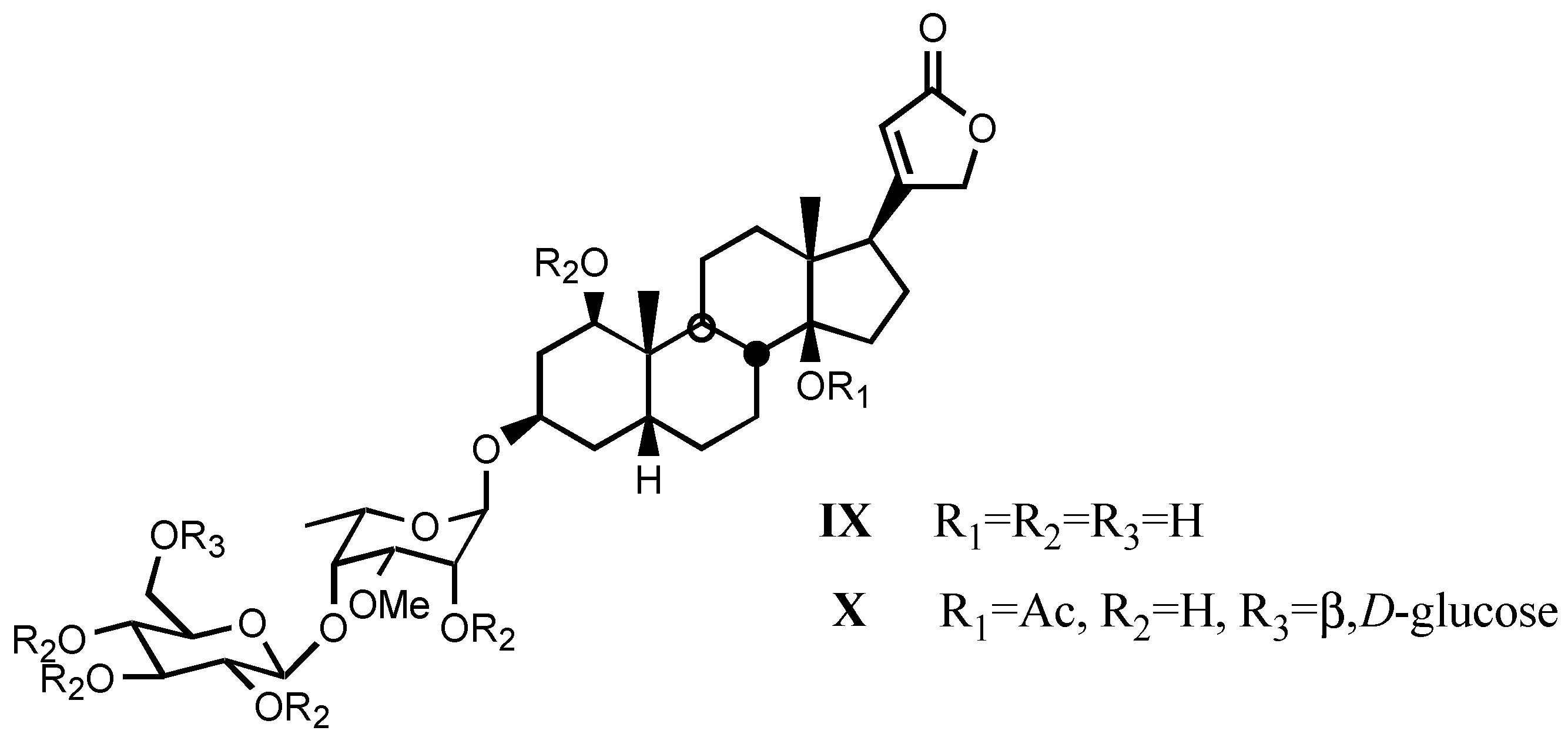
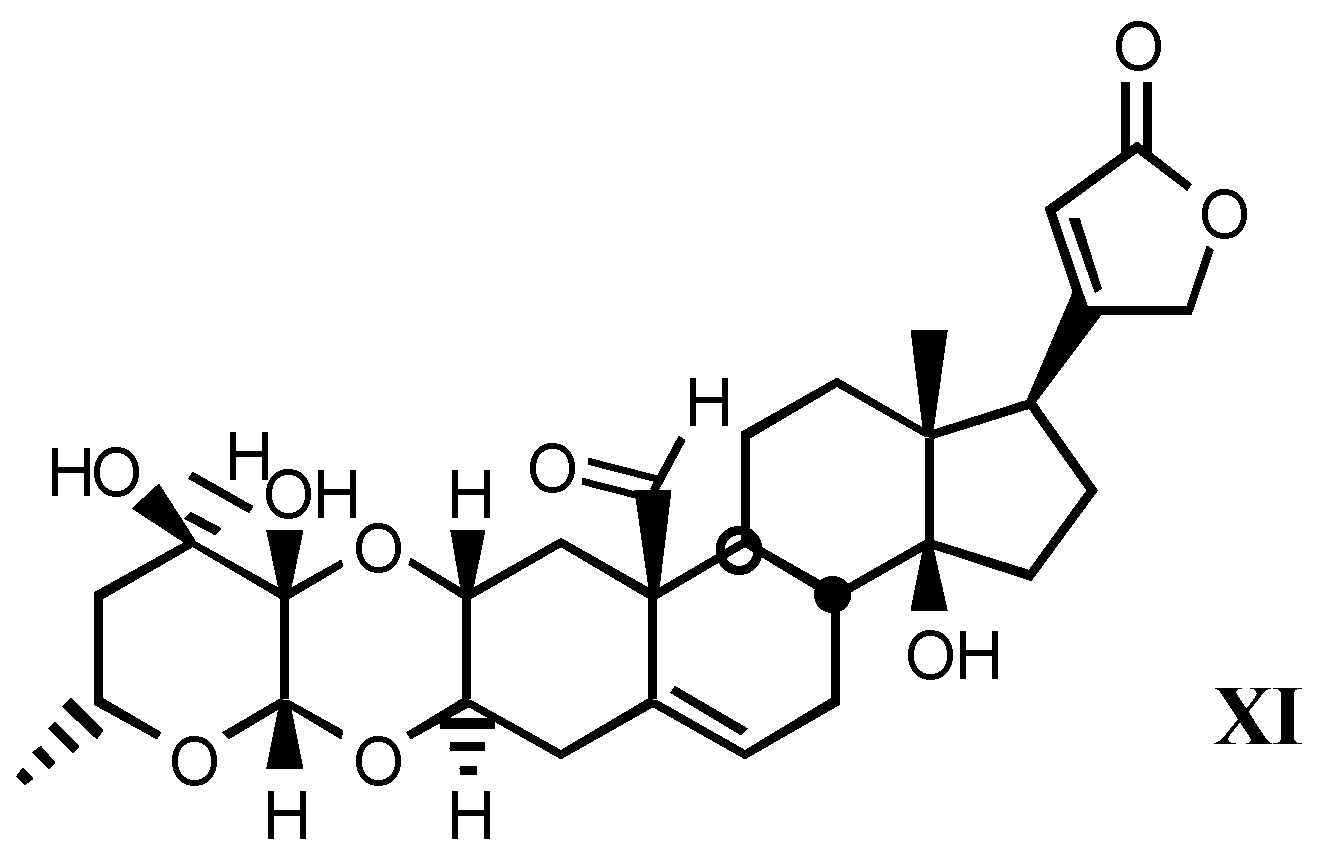
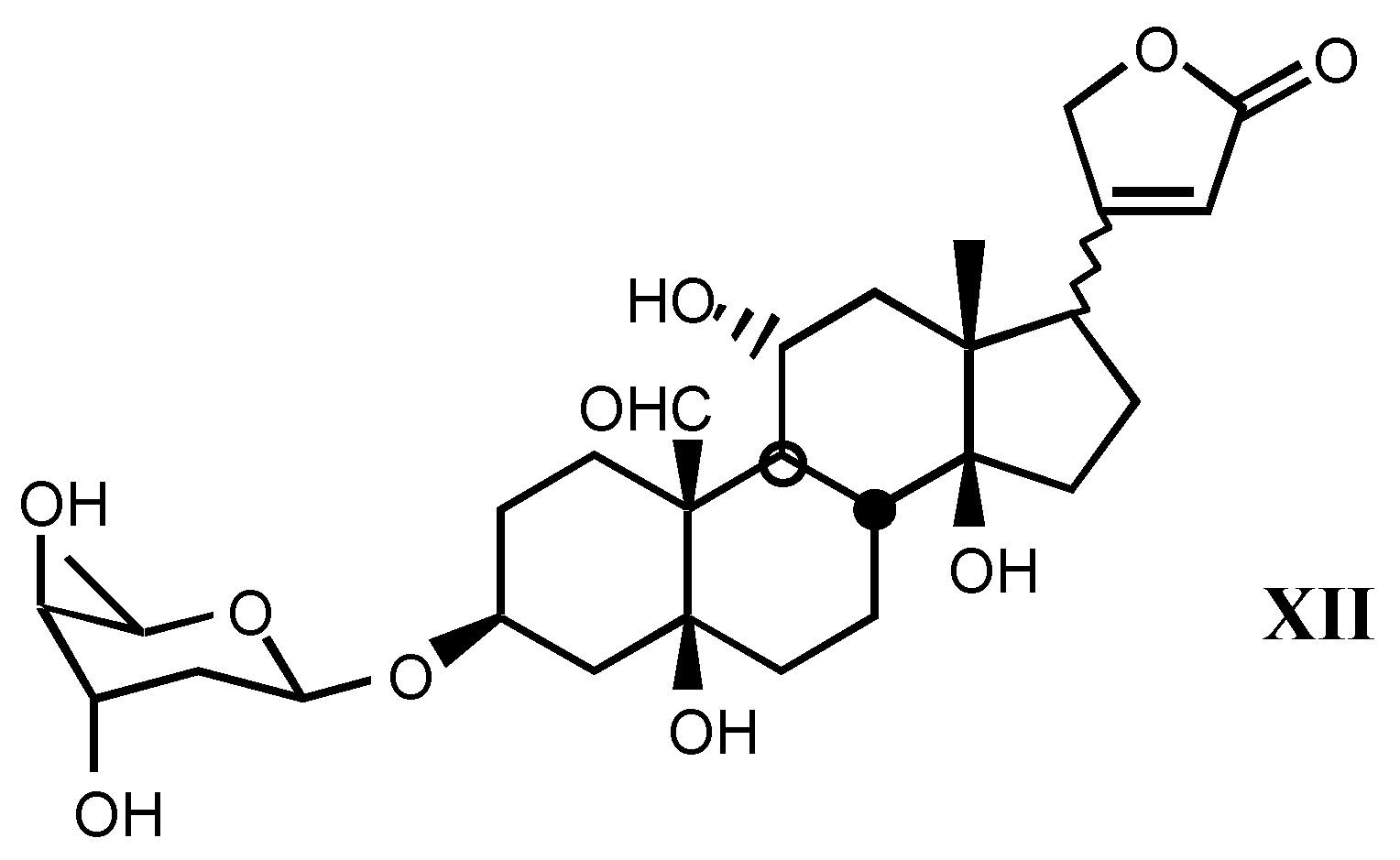
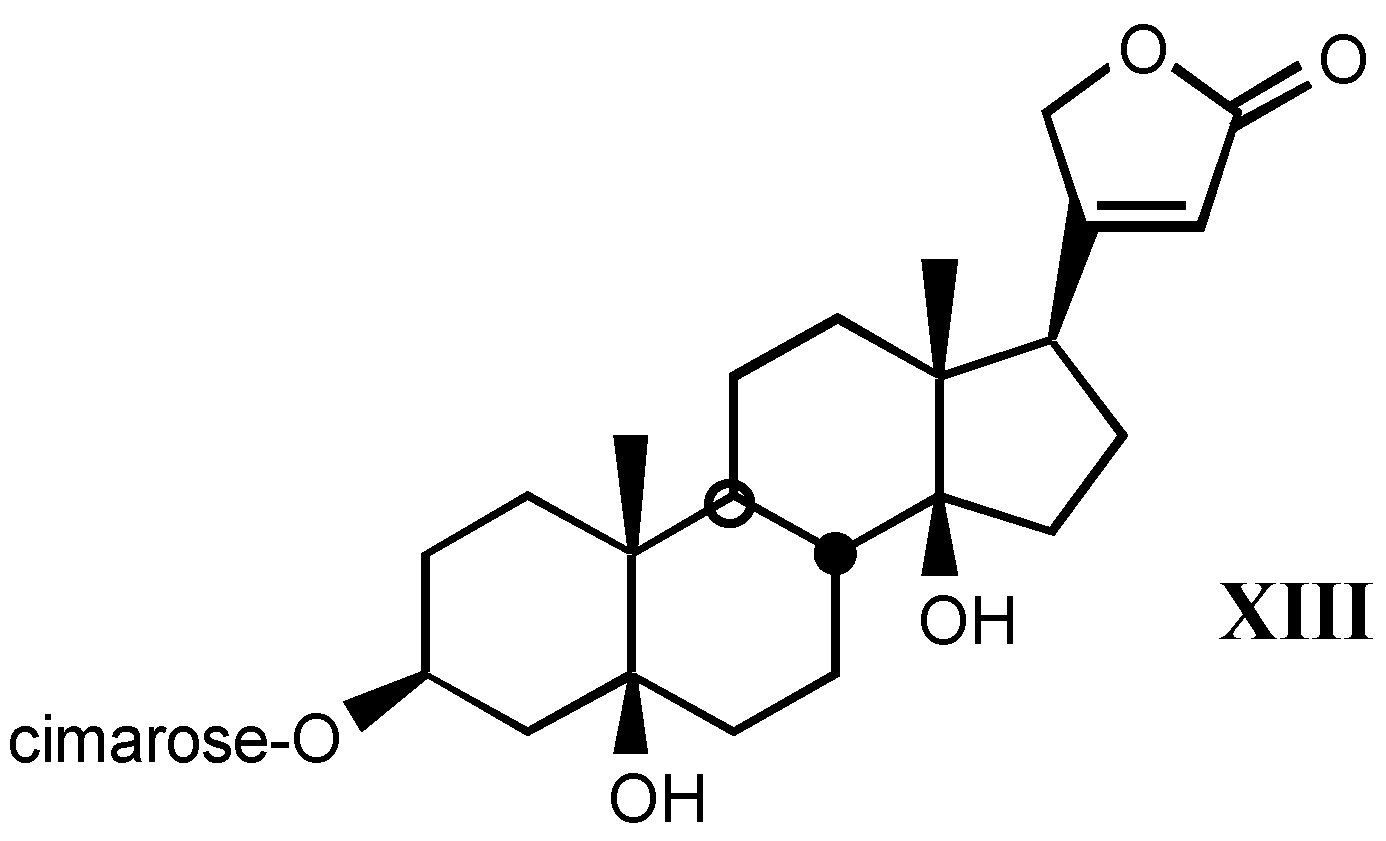
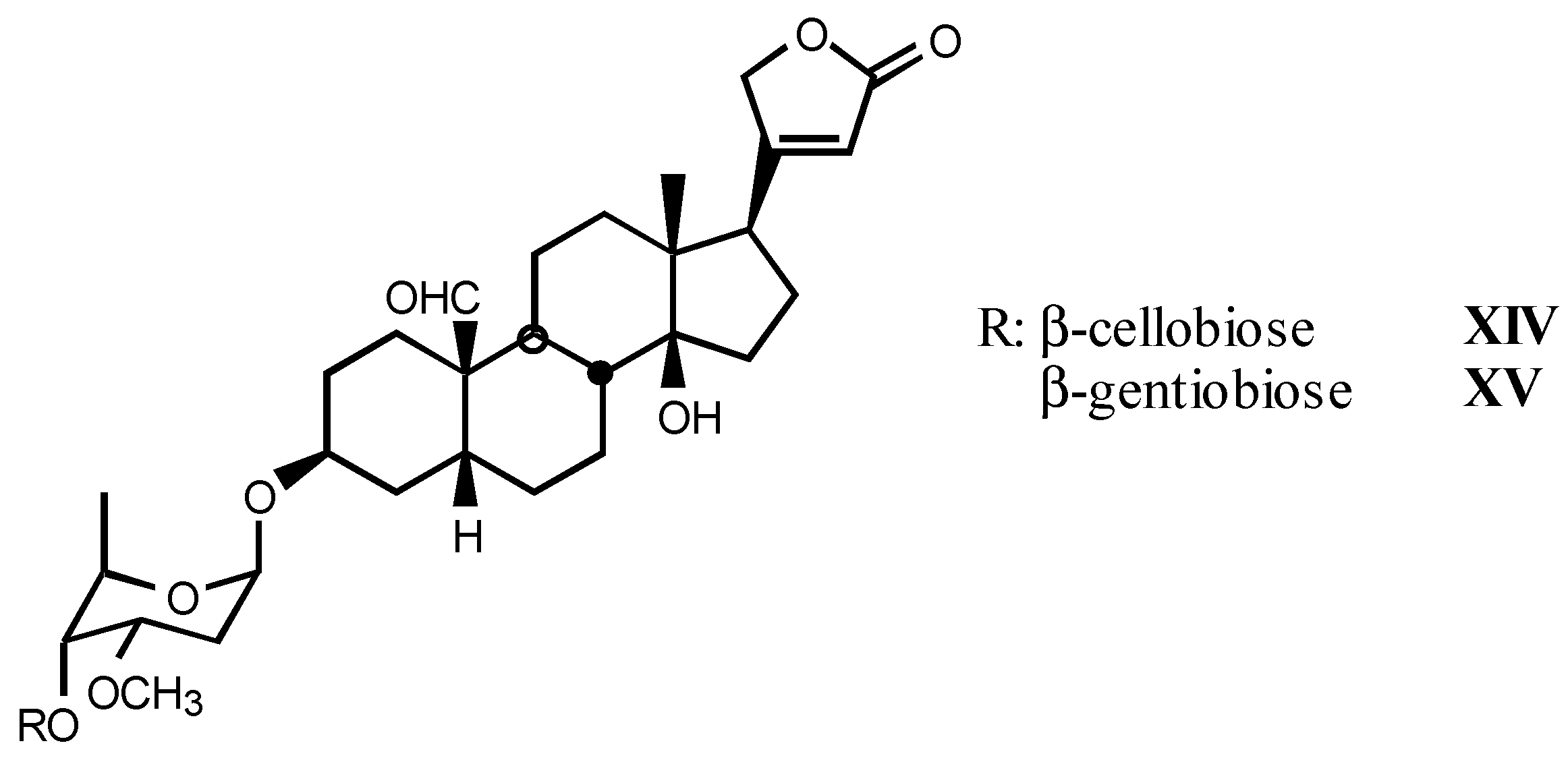
3.1.3. Natural digitalis-like glycosides recently isolated






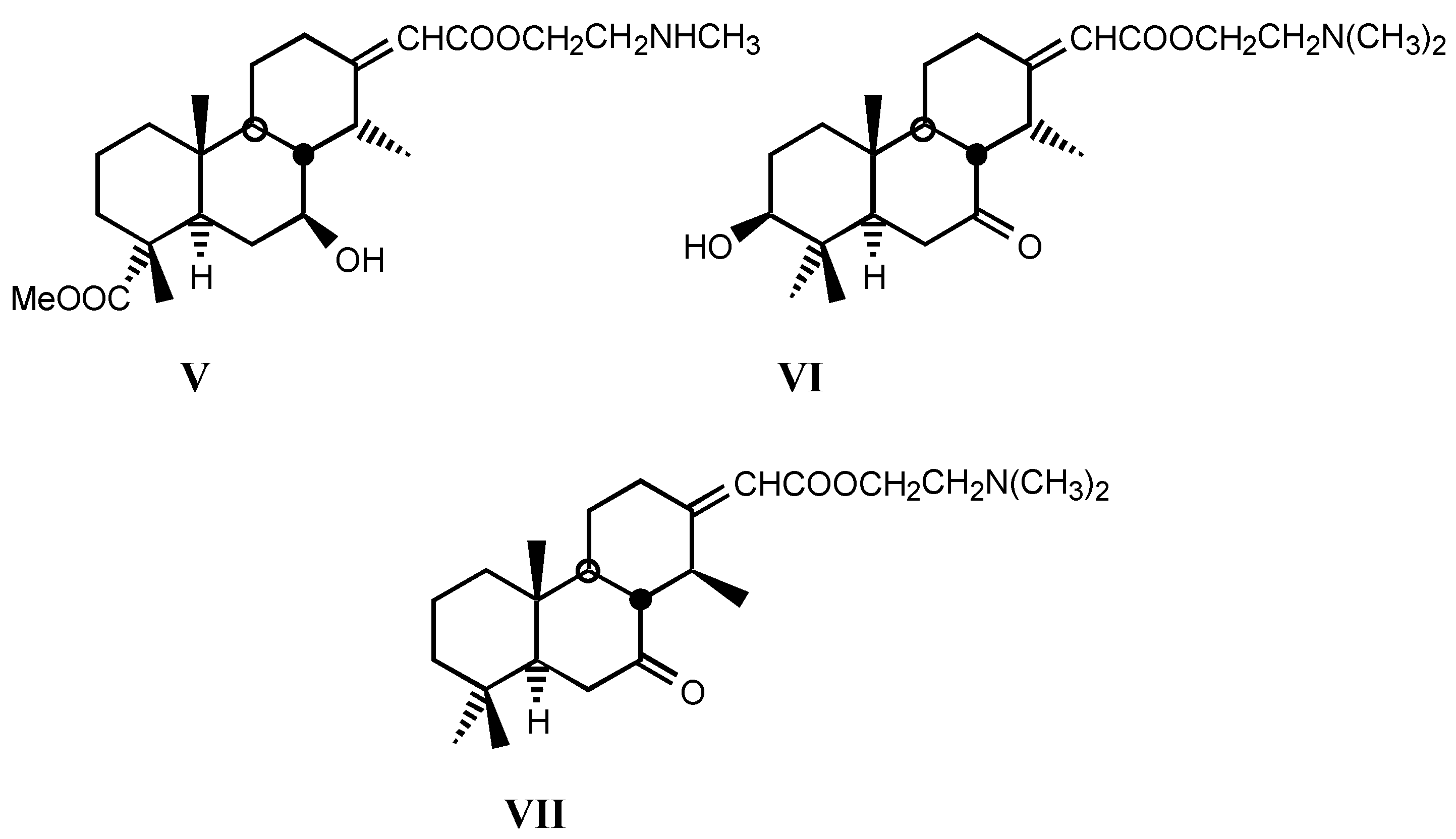
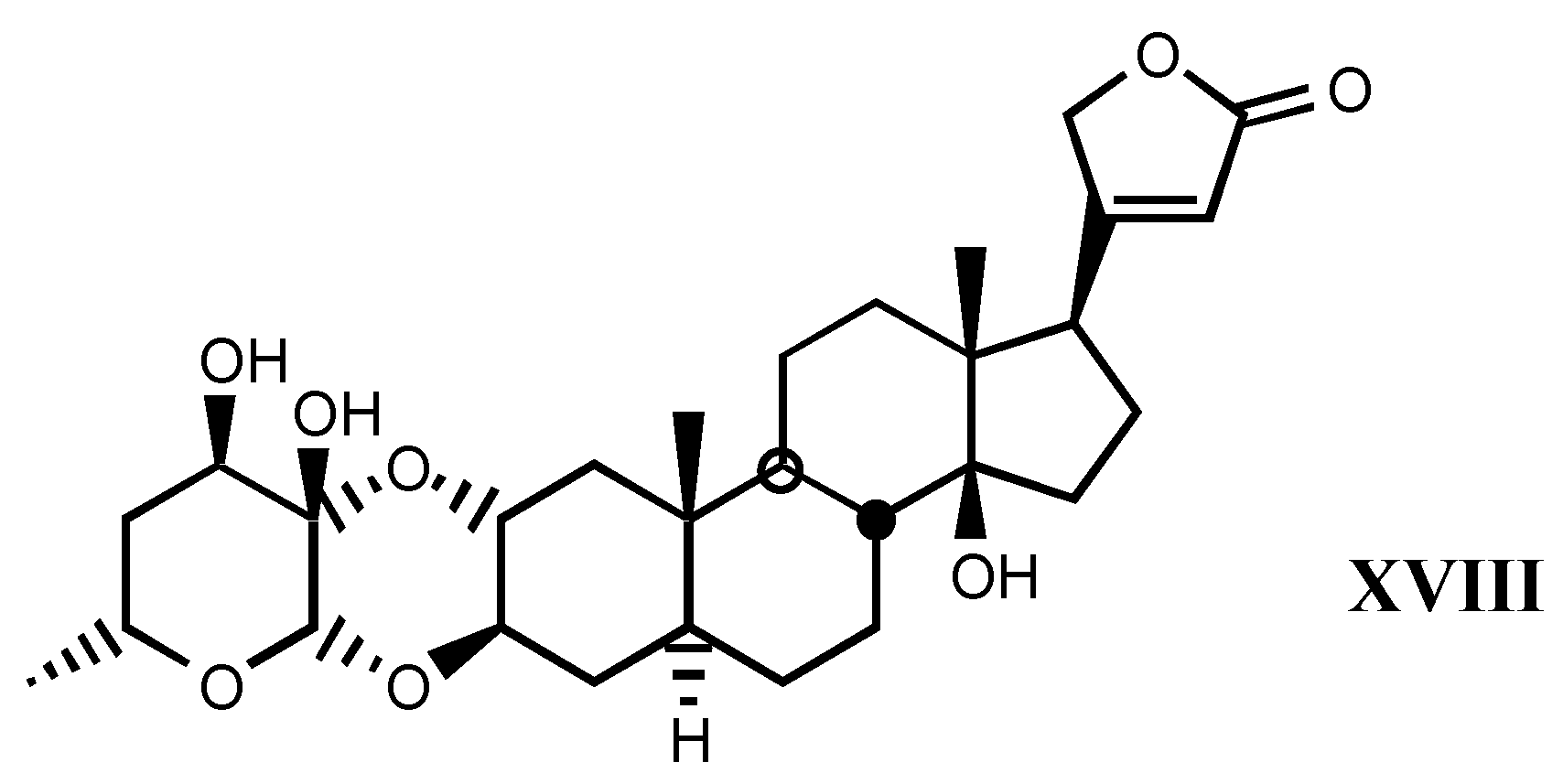
3.2. Semisynthetic and synthetic digitalis-like compounds
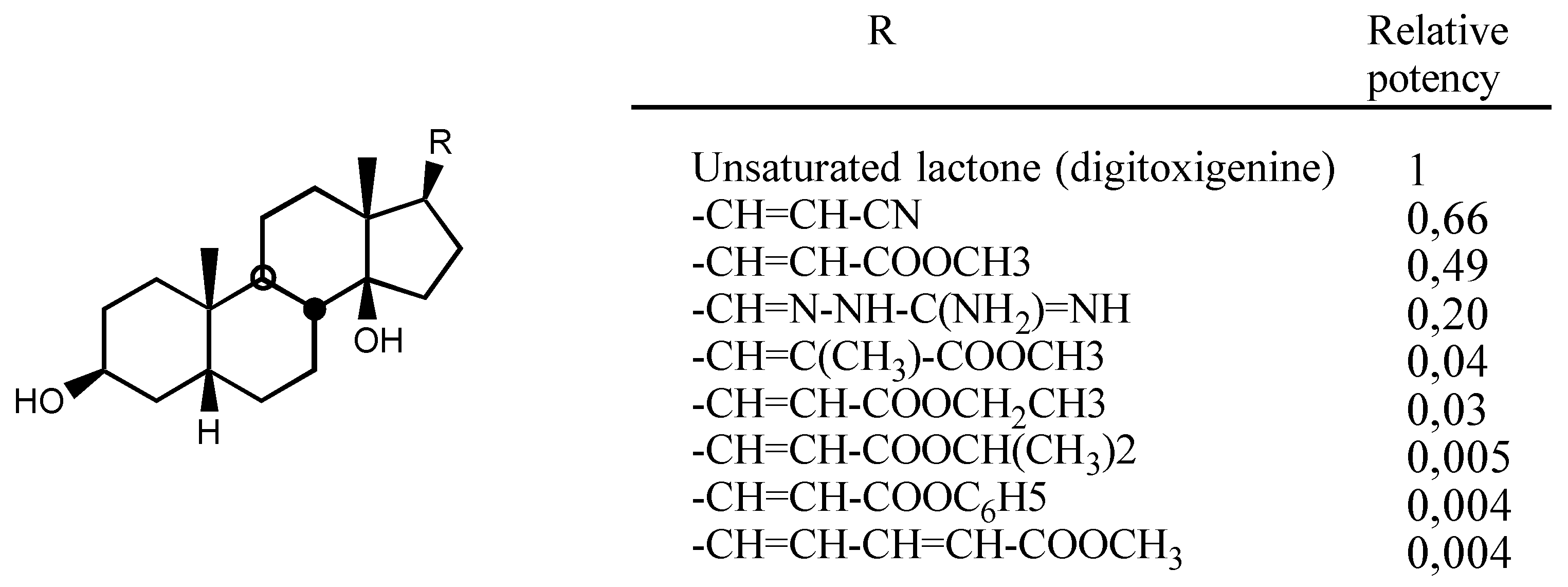
3.2.1. Structure-activity relationships (SAR)
- -
- A first one for the interaction with the steroidal framework, through hydrophobic binding.
- -
- A second one for interaction with the lactone ring showing two binding points, an electrostatic in-teraction through the electron-deficient β-Carbon atom and a Hydrogen bond to the carbonylic Oxygen atom.
- -
- A third one for interaction with sugar: hydrophobic binding through C5' and Hydrogen bond to C3'-OH.
- -
- A forth one, from which, Hydrogen bonds to the β face of the molecule could be established, in the case of having such groups.
3.2.2. Steroidal framework
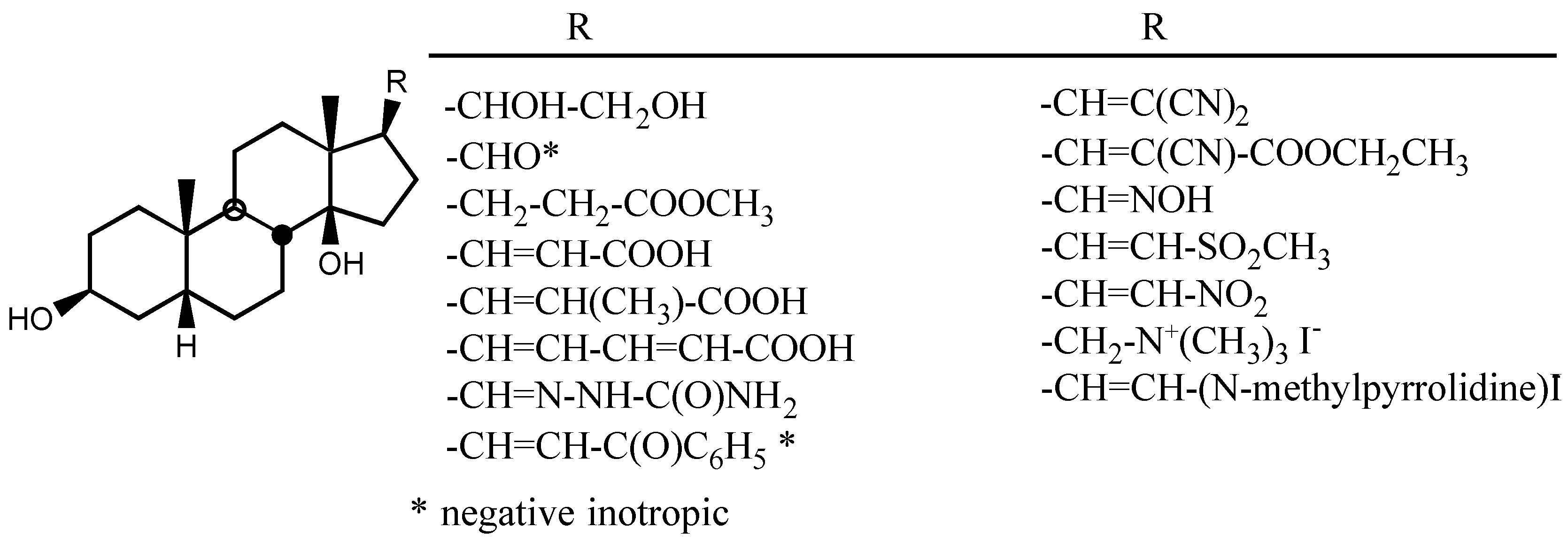
3.2.3. Side chain at C17

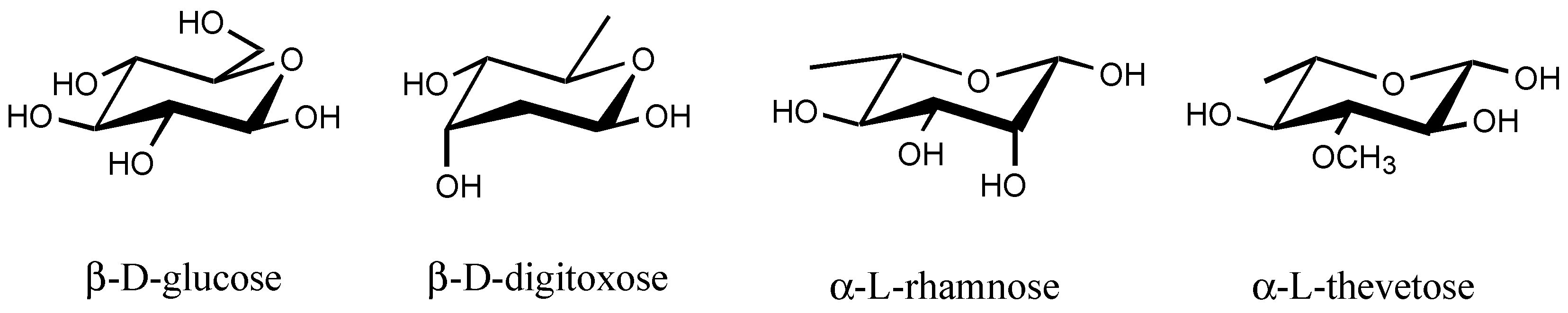
3.2.4. Sugar

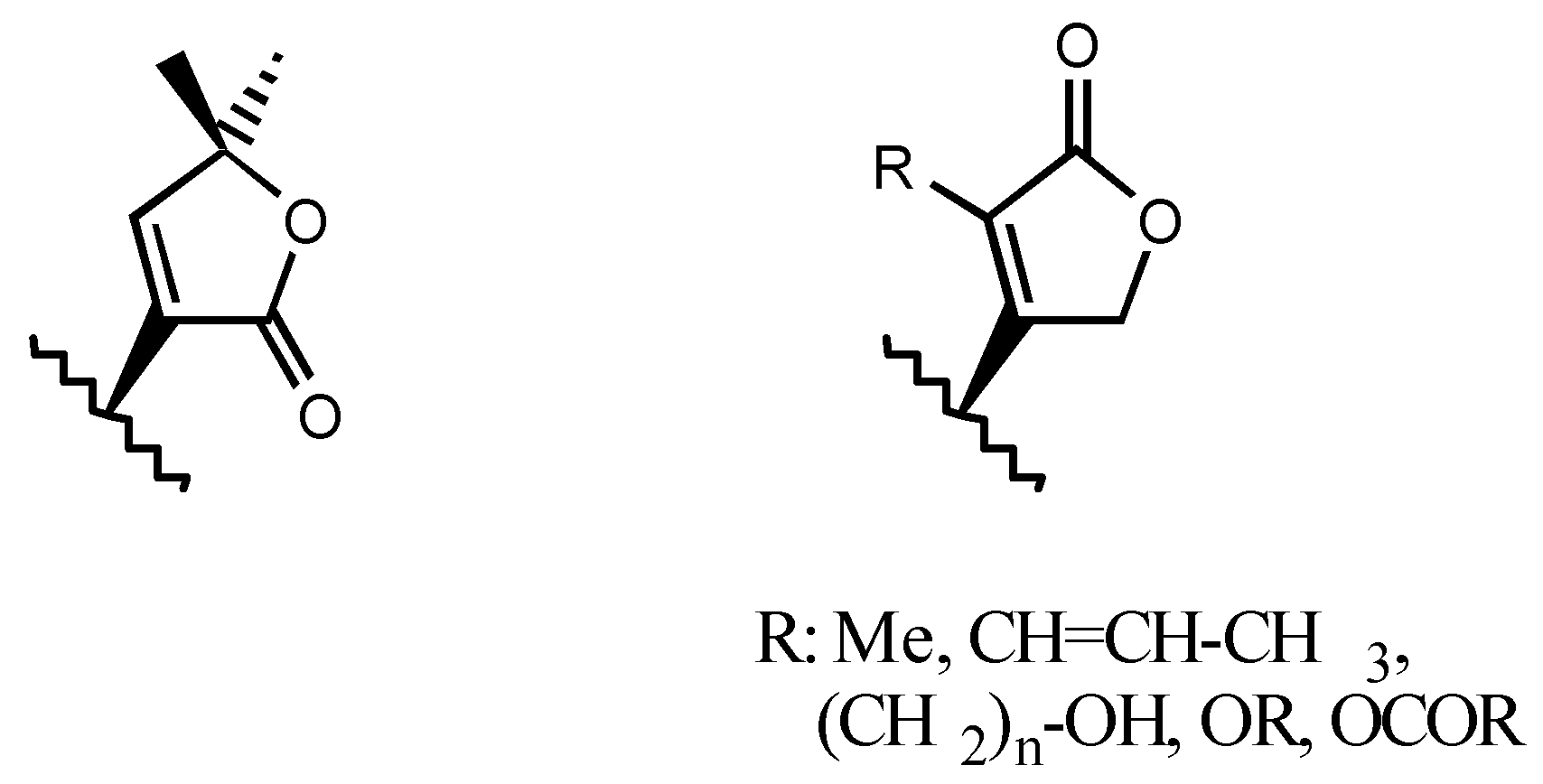
3.2.5. Other substituents at position C3
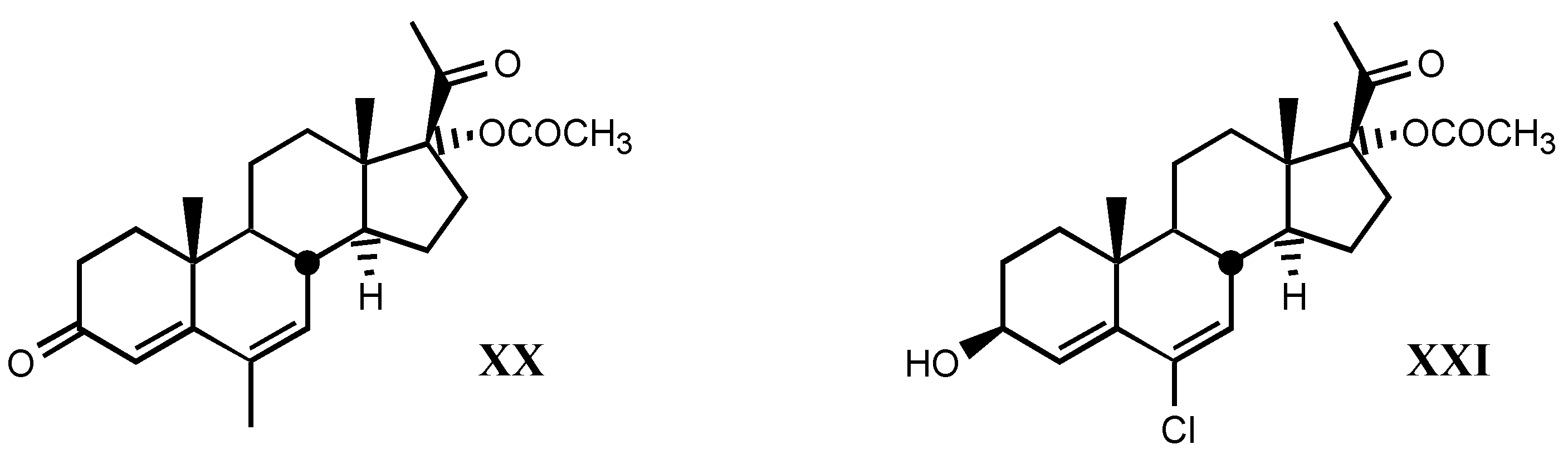
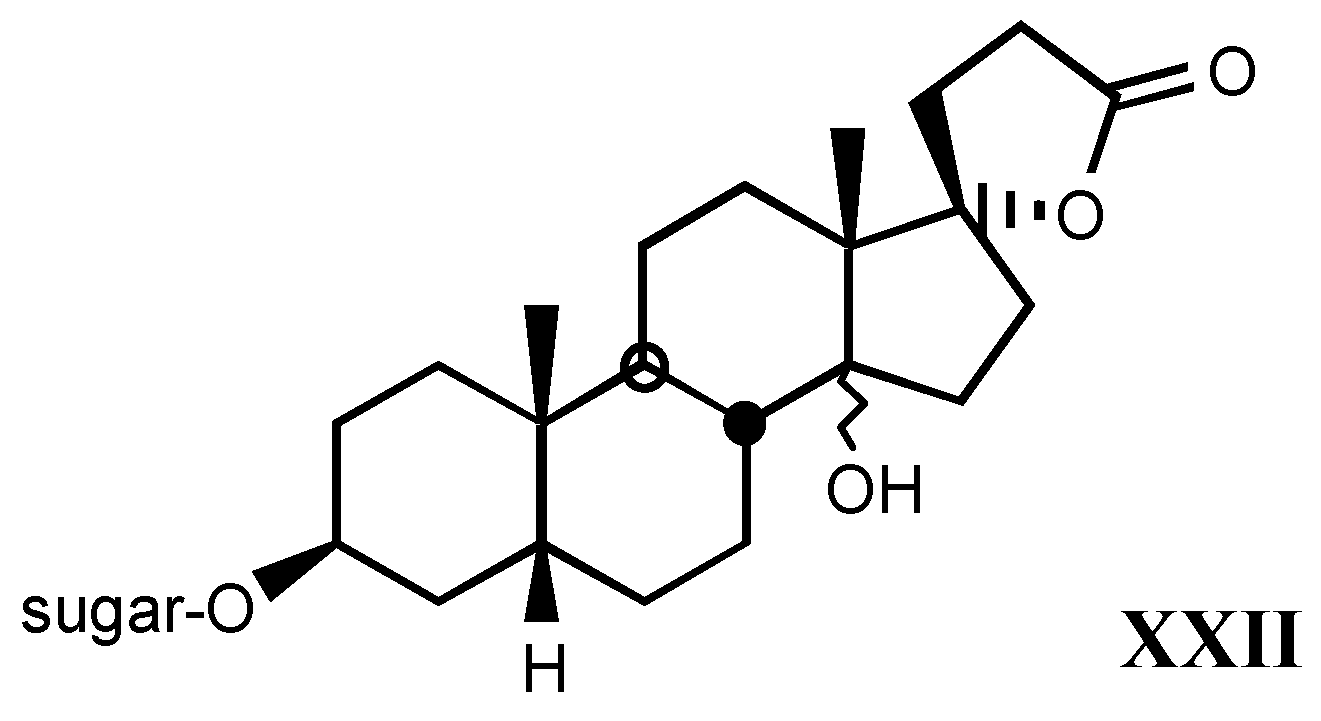
3.2.6. Pregnane derivatives




3.3. Endogenous digitalis-like factors
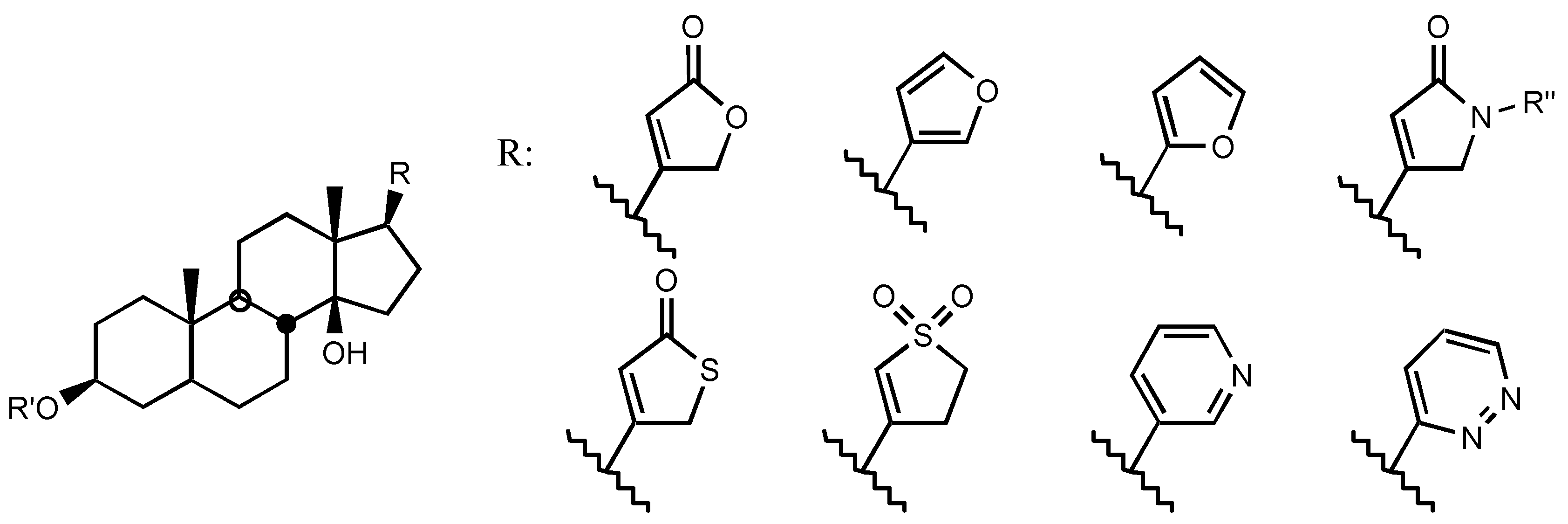
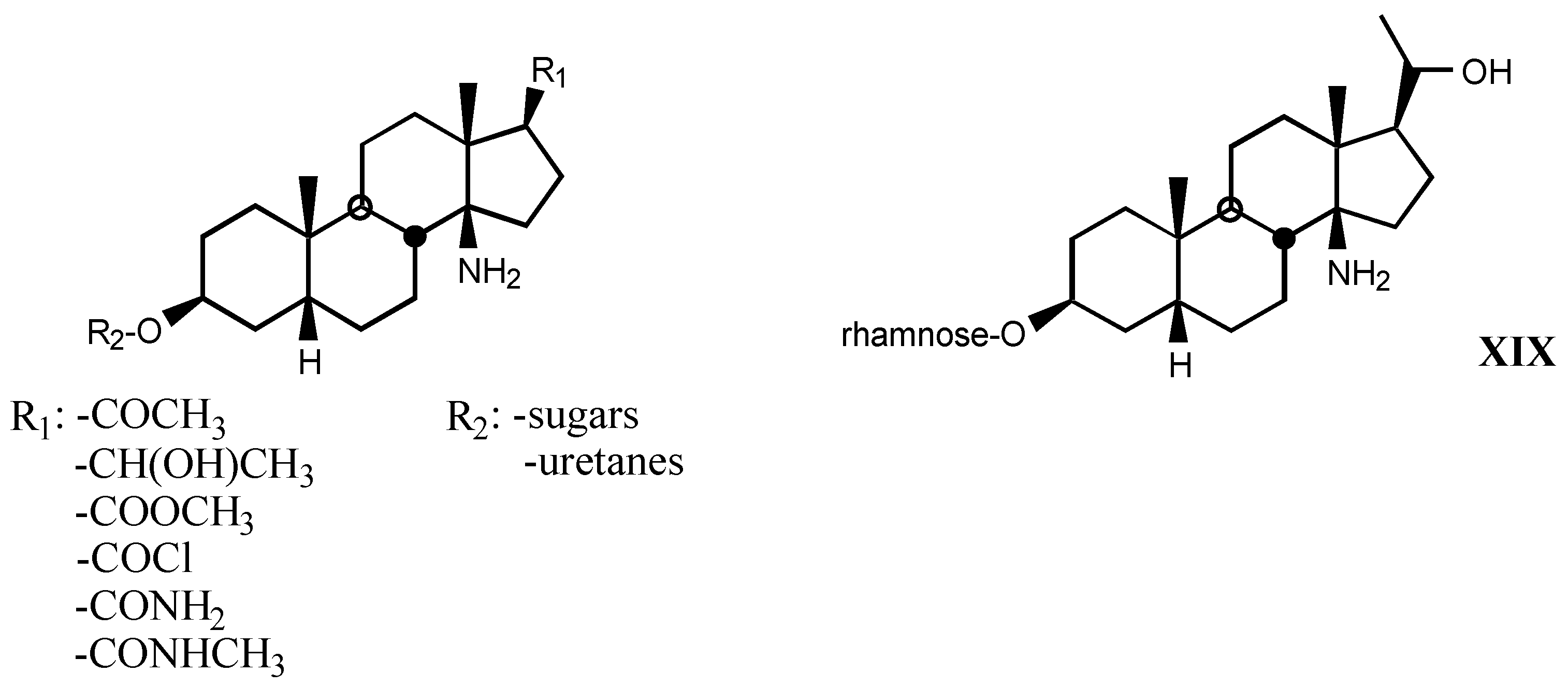
4. Digitalis Analogues Bearing Aminoguanidine Moieties
4.1. Aminoguanidine moiety in drugs
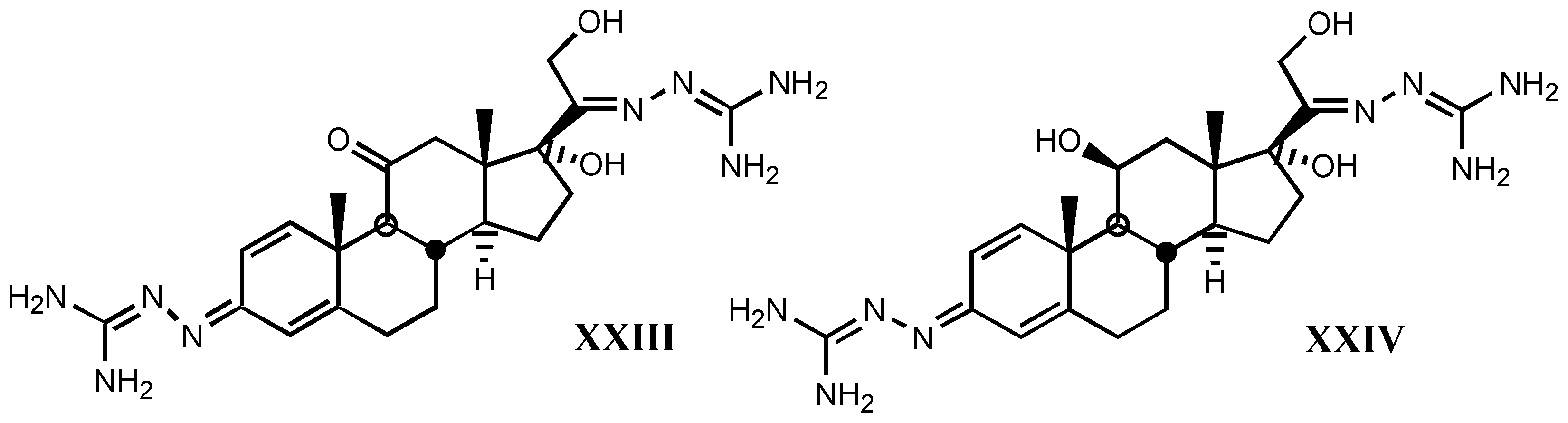
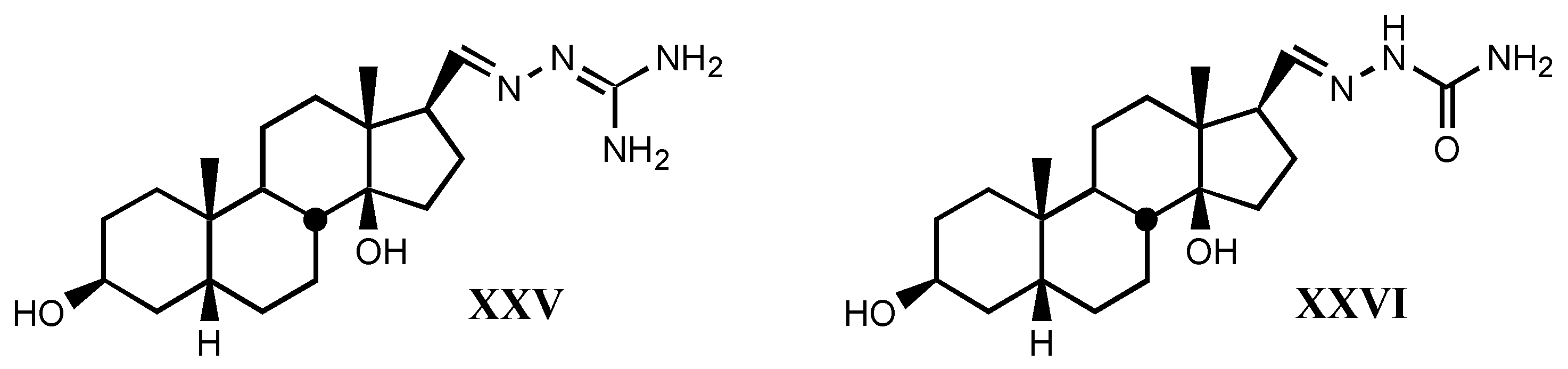
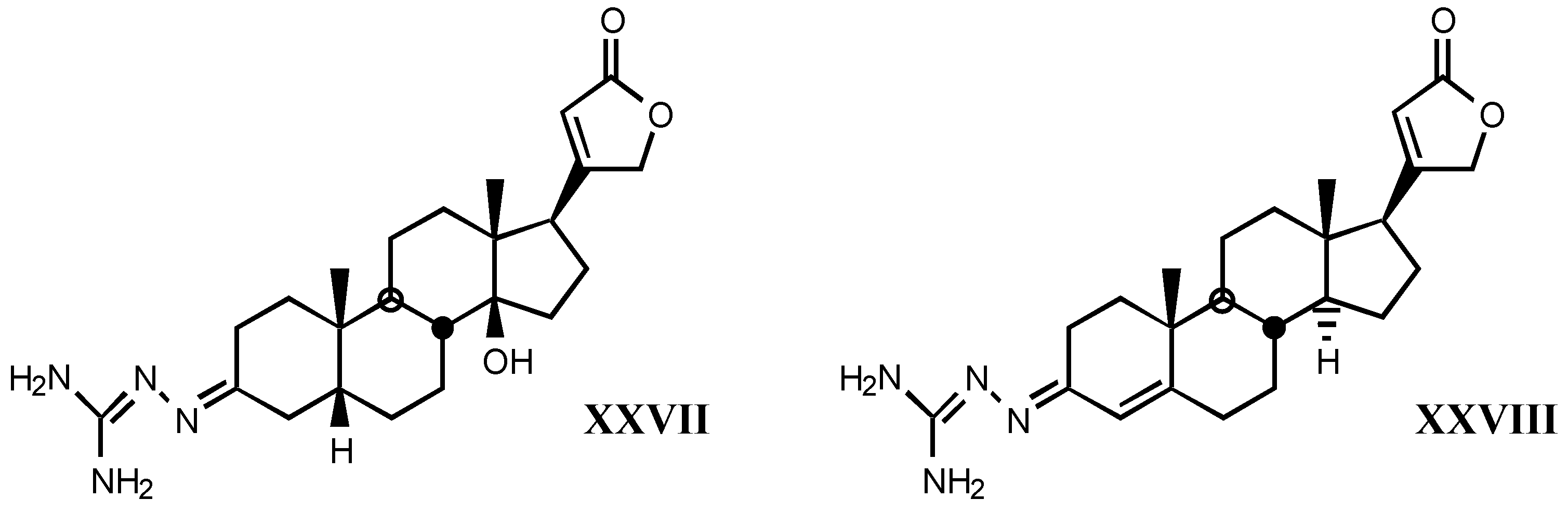
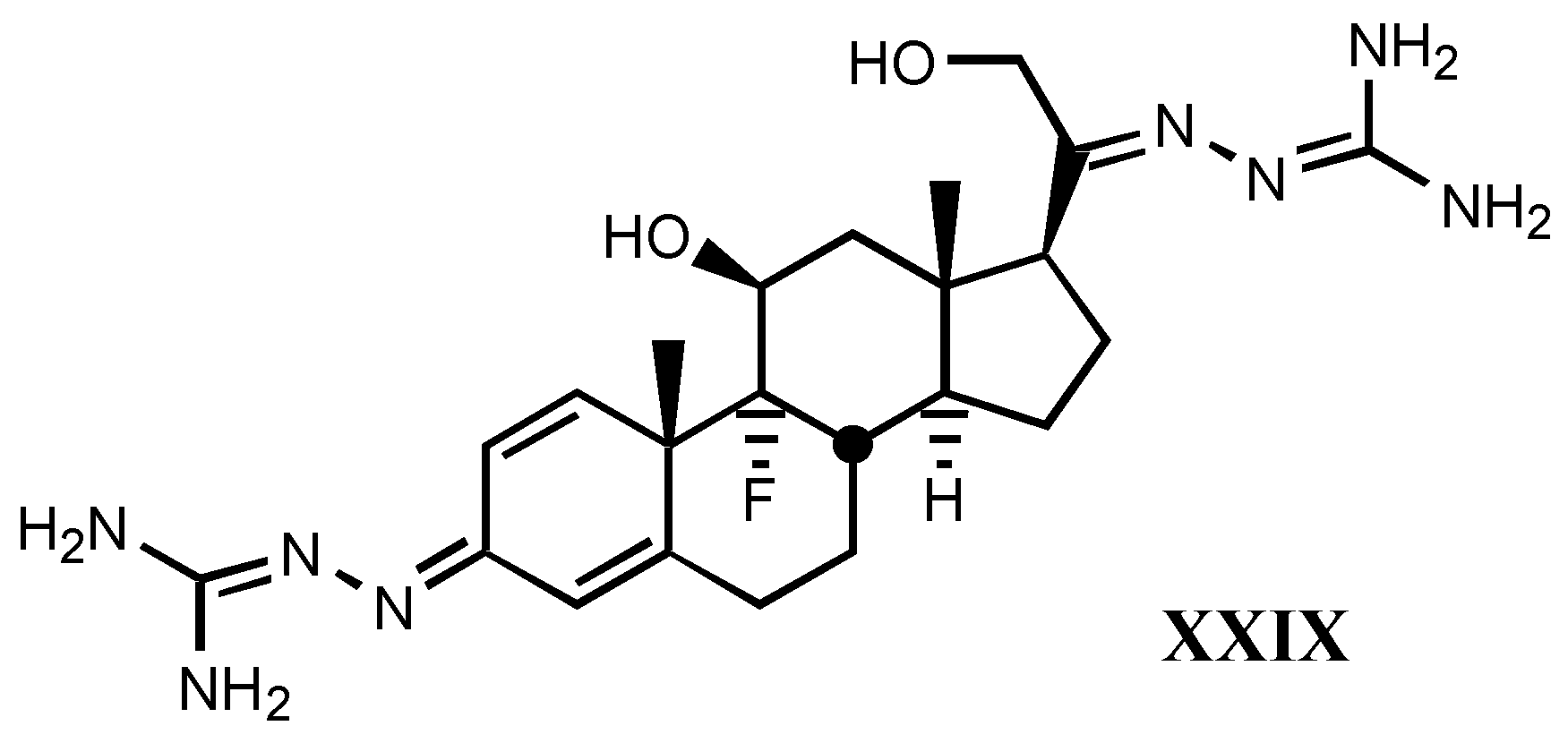
4.2. Aminoguanidines in digitalis compounds




- *
- A good correlation is observed between receptor binding and Van der Waals volume and molar refractivity of different fragments including guanylhydrazone or derivatives of it at-tached to position C17β of digitalis compounds, as well as between such a binding and pKa and PA (proton affinity) values. The two later parameters are the most important related to the affinity showed by these compounds.
- *
- The positive charge borne by guanylhydrazone (or derivatives) moiety is a very significant factor. The more protonated it is, the higher is the affinity. This one also rises with basicity.
- *
- Increasing of substituent bulk, especially when it is accompanied by an increase in the delocalization of π−electrons, produces a decreased affinity, indicating that either the re-ceptor volume for this fragment is small, or decreasing of substituent bulk allows stronger electrostatic interactions between enzyme carboxylate and the protonated hydrazone of li-gand.
Acknowledgements
References and Notes
- Withering, W. An Account of the Foxglove and Some of its Medical Uses; M. Swynney: London, 1785. [Google Scholar]
- Ferriar, J. An Essay on the Medical Properties of the Digitalis purpurea or foxglove; Sowler and Russell: Manchester, 1799. [Google Scholar]
- Haack, E.; Kaiser, F.; Gube, M.; Springler, H. Arzneim. Forsch. 1956, 6, 176–182.
- Although lactone rings of cardenolides and bufadienolides, γ-crotonolactone and α-pirone, easily react with thiol groups, Michael addition reactions to the butenolide ring of cardenolides have not been observed.
- Repke, K.R.H.; Schönfeld, W.; Weiland, J.; Megges, R.; Hache, A. Design of Enzyme Inhibi-tors as Drugs; Sandler, M., Smith, H.J., Eds.; Oxford University Press: Oxford, 1989; pp. 435–502. [Google Scholar]
- Thomas, R.E; Gray, P.; Andrews, J. Advances in Drug Research; Testa, L.B., Ed.; Academic Press: London, 1990; Vol. 19, pp. 313–575. [Google Scholar]
- Blaustein, M.P. Rev. Physiol. Biochem. Pharmacol. 1974, 70, 33–82. [PubMed]
- Skou, J.C. Biochim. Biophys. Acta 1957, 23, 394–401. [PubMed]
- Jorgensen, P.L.; Andersen, J.P. J. Membr. Biol. 1988, 103, 95–120. Modyanov, N.; Lut-senko, S.; Chertova, E.; Efremov, R.; Gulyaev, E. Acta Physiol. Scand. 1992, 146, 49–58.
- Skou, J.C. Ann. N. Y. Acad. Sci. 1982, 402, 169–184. [PubMed]
- Heijne, G.V.; Gavel, Y. Eur. J. Biochem. 1988, 174, 671–678.
- Kirley, T.L. J. Biol. Chem. 1989, 264, 7185–7192.
- Geering, K. FEBS 1991, 285, 189–193.
- Pedemonte, C.H.; Kaplan, J.H. Biochemistry 1992, 31, 10465–10470. [PubMed]
- Béguin, P.; Wang, X.; Firsov, D.; Puoti, A.; Claeys, D.; Horisberger, J.D.; Geering, K. EMBO J. 1997, 16, 4250–4260. Minor, N.T.; Sha, Q.; Nichols, C.G.; Mercer, R.W. Proc. Natl. Acad. Sci USA 1998, 95, 6521–6525. [PubMed]
- Collins, J.H.; Leszyk, J. Biochemistry 1987, 26, 8665–8668. [PubMed]Mercer, R.W.; Biemesderfer, D.; Bliss, D.P., Jr.; Collins, J.H.; Forbush, B., III. J. Cell Biol. 1993, 121, 579–586. [PubMed]
- Cornelius, F.; Skou, J.C. Biochim. Biophys. Acta 1988, 944, 223–232. [PubMed]
- Scheiner-Bobis, G.; Fahlbusch, K.; Schoner, W. J. Biochem. 1987, 168, 123–131.
- Scheiner-Bobis, G.; Buxbaum, E.; Schoner, W. The Na+,K+-Pump, Part A: Molecular Aspects; Skou, J.C., Nørby, J.G., Maunsbach, A.B., Esmann, M., Eds.; Alan R. Liss: New York, 1988; p. 219. [Google Scholar]
- Skriver, E.; Kaveus, U.; Hebert, H.; Maunsbach, A.B. J. Struct. Biol. 1992, 108, 176–185. [PubMed]Chetverin, A.B. EBS Lett. 1986, 196, 121–125.
- Na+ and K+ concentrations at rest are: [Na+]int.=7-20 mM, [Na+]ext.=140 mM, [K+]int.=110-120 mM, [K+]ext.= 4-5 mM.
- Albers, R.W. Annu. Rev. Biochem. 1967, 36, 727–756. [PubMed]
- Post, R.L.; Kume, S.; Tobin, T.; Orcutt, T.; Sen, A.K. J. Gen. Physiol. 1969, 54, 306S–326S.
- Shull, G.E.; Schwartz, A.; Kingrel, J.B. Nature 1985, 316, 691–695. [PubMed]
- Shamraj, O.I.; Lingrel, J.B. Proc. Natl. Acad. Sci. USA 1994, 91, 12952–12956. [PubMed]
- Lingrel, J.B.; Orlowski, J.; Shull, M.M.; Price, E.M. Prog. Nucleic Acid Res. Mol. Biol. 1990, 38, 37–89. [PubMed]
- Shyjan, A.W.; Gottardi, C.; Levenson, R. J. Biol. Chem. 1990, 265, 5166–5169. [PubMed]
- Malik, N.; Canfield, V.A.; Beckers, M.C.; Gros, P.; Levenson, R. J. Biol. Chem. 1996, 271, 22754–22758. [PubMed]Yu, C.; Xie, Z.; Askari, A.; Modyanov, N.N. Arch. Biochem. Biophys. 1997, 345, 143–149.
- Good, P.J.; Richter, K.; Dawid, I.B. Proc. Natl. Acad. Sci. USA 1990, 87, 9088–9092. [PubMed]
- Shull, M.M.; Lingrel, J.B. Proc. Natl. Acad. Sci. USA 1987, 84, 4039–4043. [PubMed]Shull, M.M.; Pugh, D.G.; Lingrel, J.B. J. Biol. Chem. 1989, 264, 17532–17543. [PubMed]Shull, M.M.; Pugh, D.G.; Lingrel, J.B. Genomics 1990, 6, 451–460. [PubMed]Lane, L.K.; Shull, M.M.; Whitmer, K.R.; Lingrel, J.B. Genomics 1989, 5, 445–453. [PubMed]Yang, F.T.; Schneider, J.W.; Lindgren, V.; Shull, M.M.; Benz, E., Jr.; Lingrel, J.B.; Francke, U. Genomics 1988, 2, 128–138.
- Levenson, R. Rev. Physiol. Biochem. Pharmacol. 1994, 123, 1–45. [PubMed]
- Geering, K. Curr. Op. Nephrol. Hypertens. 1997, 6, 434–439.
- Thomas, R.E. Molecular Structure and Biological Activity of Steroids; Bohl, M., Daux, W.L., Eds.; CRC Press: Boca Raton, 1992; pp. 399–464. [Google Scholar]
- Lee, C.O. Am. J. Physiol. 1985, 249, 367–378.
- Gillis, R.A.; Quest, J.A. Cardiac Glycosides; Erdmann, E., Greeff, K., Skou, J.C., Eds.; Steinkopff: Darmstadt, 1986; pp. 347–356. [Google Scholar]
- Steyn, P.S.; van Heerden, F.R. Nat. Prod. Reports 1988, 15, 397–413.
- Fieser, L.F.; Fieser, M. Natural Products Related to Phenantrene; Reinold corp: New York, 1949; pp. 507–577. [Google Scholar] Sivadijan, J. Traité de Chimie Organique; Grignard, V., Dupont, G., Locquin, R., Eds.; Masson: Paris, 1949; pp. 1076–1079. [Google Scholar]
- Deepak, D.; Srivastava, S.; Khare, N.K.; Khare, A. Fortsch. Chem. Org. Naturst. 1996, 69, 71–155. Gaignault, J.C.; Bidet, D. Fitoterapia 1988, 59, 259–315.
- Imre, Z.; Yurdun, T. Planta Medica 1988, 54, 529–531. [PubMed]
- Pieri, F.; Arnould-Guerin, M.L.; Sefraoni, E.H. Fitoterapia 1992, 63, 333–336.
- Cheung, H.T.A.; Nelson, C.J. J. Chem. Soc. Perkin Trans. I 1989, 1563–1570.
- Gil, R.R.; Lin, L.Z.; Chai, H.B.; Pezzuto, J.M.; Cordell, G.A. J. Nat. Products 1995, 58, 848–856.
- Umehara, K.; Sumii, N.; Satoh, H.; Miyase, T.; Kuroyagani, M.; Ueno, A. Chem Pharm. Bull. 1995, 43, 1565–1568.
- Abe, F.; Yamauchi, T. Chem. Pharm. Bull. 1994, 42, 2028–2031.
- Thomas, R.E.; Gray, P.; Andrews, J. Advances in Drug Research; Testa, L.B., Ed.; Aca-demic Press: London, 1990; Vol. 19, pp. 313–575. [Google Scholar] Thomas, R.E. Burger’s Medicinal Chem-istry and Drug Discovery, 5th ed.; Wolff, M.E., Ed.; John Wiley&Sons: New York, 1996; Vol. 2, pp. 153–261. [Google Scholar] Repke, K.R.H.; Weiland, J.; Megges, R. Progress in Medicinal Chemistry; Ellis, G.P., Luscombe, D.K., Eds.; Elsevier: Amsterdam, 1993; Vol. 30, pp. 135–202. [Google Scholar] Repke, K.R.H.; Weiland, J.; Megges, R. Angew. Chem. Int. Ed. Engl. 1995, 34, 282–294. Bohl, M.; Süssmilch, R. Eur. J. Med. Chem. 1986, 21, 193–198. Repke, K.R.H.; Weiland, J.; Megges, R.; Schon, R. J. Enzyme Inhib. 1996, 10, 147–157. [PubMed]
- Schönfeld, W.; Weiland, J.; Lindig, C.; Masnyk, M.; Kabat, M.M.; Kurek, A.; Wicha, J.; Repke, K.R.H. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1985, 329, 414–426. Repke, K.R.H. Trends Pharmacol. Sci. 1985, 6, 275–278.
- Burt, S.K.; Mackay, D.; Hagler, A.T. Computer-Aided Drug Design: Methods and Applica-tions; Perun, T.J., Propts, C.L., Eds.; Marcel Dekker: New York, 1989; pp. 55–91. [Google Scholar]
- Schönfeld, W.; Schönfeld, R.; Menke, K.H.; Weiland, J.; Repke, K.R.H. Biochem. Pharmacol. 1986, 35, 3221–3231.
- Shigei, T.; Tsuru, H.; Saito, Y.; Okada, M. Experientia 1973, 29, 449–450. [PubMed]
- Griffin, J.F.; Rohrer, D.C.; Ahmed, K.; From, A.H.L.; Hashimoto, T.; Rathore, H.; Fullerton, D.S. Mol. Pharmacol. 1986, 29, 270–274. [PubMed]Hashimoto, T.; Rathore, H.; Satoh, D.; Hong, G.; Griffin, J.F.; From, A.H.L.; Ahmed, K.; Fullerton, D.S. J. Med. Chem. 1986, 29, 997–1003. [PubMed]
- Gobbini, M.; Benicchio, A.; Marazzi, G.; Padoani, G.; Torri, M.; Melloni, P. Steroids 1996, 61, 572–582. [PubMed]
- San Feliciano, A.; Medarde, M.; Caballero, E.; Hebrero, B.; Tomé, F. Tetrahedron 1990, 46, 6789–6798.
- Medarde, M.; Caballero, E.; Tomé, F.; Gracia, P.G.; Boya, M.; San Feliciano, A. Synth. Com-mun. 1995, 25, 1377–1382. Medarde, M.; Tomé, F.; López, J.L.; Caballero, E.; Boya, M.; Melero, C.P.; San Feliciano, A. Tetrahedron Lett. 1994, 35, 8683–8686. Medarde, M.; Cabal-lero, E.; Melero, C.P.; Tomé, F.; San Feliciano, A. Tetrahedron: Asymmetry 1997, 8, 2075–2077.
- Melero, C.P. Ph.D. disertation, Universidad de Salamanca, 1999.
- Melero, C.P.; Sevillano, L.G.; Caballero, E.; Tomé, F.; Carrón, R.; Montero, M.J.; San Fe-liciano, A.; Medarde, M. Bioorg. Med. Chem. Lett. 1998, 8, 3217–3222. [PubMed]Sevillano, L.G.; Melero, C.P.; Boya, M.; López, J.L.; Tomé, F.; Caballero, E.; Carrón, R.; Montero, M.J.; Medarde, M.; San Feliciano, A. Bioorg. Med. Chem. 1999, (in press).
- Smith, P.; Brown, L.; Boutagy, J.; Thomas, R.E. J. Med. Chem. 1982, 25, 1222–1226. [PubMed]
- Schönfeld, W.; Repke, K.R.H. Quant. Struct.-Act. Relat. 1988, 7, 160–165.
- Kahn, J.B.; Acheson, G.H. J. Pharmacol. Exp. Ther. 1955, 115, 301–318. Guzmán, A.; Muchowski, J.M.; Strosberg, A.M.; Sims, J.M. Can J. Chem. 1981, 59, 3241–3247. San Fe-liciano, A.; Medarde, M.; Caballero, C.; Hebrero, M.B.; Tomé, F.; Prieto, P.; Montero, M.J. Eur. J. Med. Chem. 1990, 25, 413–417. Medarde, M.; Caballero, E.; Tomé, F.; García, A.; Montero, M.J.; Carrón, R.; San Feliciano, A. Eur. J. Med. Chem. 1993, 28, 887–892.
- Bohl, M.; Süssmilch, R. Eur. J. Med. Chem., Chim. Ther. 1986, 21, 193–198.
- Boobbyer, D.N.A.; Goodford, P.J.; McWhinnie, P.M.; Wade, R.C. J. Med. Chem. 1989, 32, 1083–1094. [PubMed]
- Pastelin, G.; Méndez, R. Life Sci. 1983, 32, 1905–1909. [PubMed]Staroske, T.; Henning, L.; Welzel, P.; Hofmann, H.J.; Müller, D.; Haüsler, T.; Sheldrick, W.S.; Zillikens, S.; Gretzer, B.; Pusch, H.; Glitsch, H.G. Tetrahedron 1996, 52, 12723–12744.
- Fullerton, D.S.; Yoshioka, K.; Rohrer, D.C.; From, A.H.L.; Ahmed, K. Science 1979, 205, 917–919. [PubMed]From, A.H.L.; Fullerton, D.S.; Deffo, T.; Kitatsuji, E.; Rohrer, D.C.; Ahmed, A. J. Mol. Cell. Cardiol. 1984, 16, 835–842. [PubMed]
- Fullerton, D.S.; Yoshioka, K.; Rohrer, D.C.; From, A.H.L.; Ahmed, K. Mol. Pharmacol. 1980, 17, 43–51. [PubMed]
- Fullerton, D.S.; Ahmed, K.; From, A.H.L.; McParland, R.H.; Rohrer, D.C.; Griffin, J.F. Mo-lecular Graphics and Drug Design; Burgen, A.S.V., Roberts, G.C.K., Tute, M.S., Eds.; Elsevier: Amsterdam, 1986; pp. 257–284. [Google Scholar]
- Scrocco, E.; Tomasi, J. Adv. Quantum Chem. 1978, 11, 115–194.
- Theil, F.; Lindig, C.; Repke, K.R.H. J. Prakt. Chem. 1980, 322, 1012–1020.
- Thomas, R.E.; Boutagy, J.; Gelbart, A. J. Pharm. Exp. Ther. 1974, 191, 219–231.
- Fullerton, D.S.; Yoshioka, K.; Rohrer, D.C.; From, A.H.L.; Ahmed, K. J. Med. Chem. 1979, 22, 529–533. [PubMed]
- Akera, T. Cardiac Glycosydes, Part I, Experimental Pharmacology; Greeff, K., Ed.; Springer: Berlin, 1981; Vol. 56, I, pp. 287–336. [Google Scholar]
- Chiu, F.C.K.; Watson, R.T. J. Med. Chem. 1985, 28, 509–515. [PubMed]Yoda, A. Mol. Pharmacol. 1973, 9, 51–60. [PubMed]
- Fullerton, D.S.; Kihara, M.; Deffo, T.; Kitatsuji, E.; Ahmed, K.; Simat, B.; From, A.H.L.; Rohrer, D.C. J. Med. Chem. 1984, 27, 256–261. [PubMed]
- Rathore, H.; From, A.H.L.; Ahmed, K.; Fullerton, D.S. J. Med. Chem. 1986, 29, 1945–1952. [PubMed]
- Brown, L.; Thomas, R.E. Arzneim. Forsch. 1983, 33, 814–817.
- Randimbivololona, F.; Pellegrin, P.; Lesne, M. J. Pharm. Belg. 1984, 39, 225–232. [PubMed]
- From, A.H.L.; Fullerton, D.S.; Ahmed, K. Mol. Cell Biochem. 1990, 94, 157–165. [PubMed]
- Gobbini, M.; Benicchio, A.; Padoani, G.; Torri, M.; Melloni, P. Biorg. Med. Chem. Lett. 1997, 7, 469–472.
- Kyte, J. J. Biol. Chem. 1972, 247, 7634–7641. [PubMed]
- Jarreau, F.X.; Koening, J.J. European Patent EP 3455, 1979.
- Ippolito, J.A.; Alexander, R.S.; Christianson, D.W. J. Mol. Biol. 1990, 215, 457–471.
- Hansen, O. Pharmacol. Rev. 1984, 36, 143–163. [PubMed]
- Sakakibara, M.; Uchida, A.O. Biosci. Biotech. Biochemistry 1996, 60, 405–410. Sakakibara, M.; Uchida, A.O. Biosci. Biotech. Biochemistry 1996, 60, 411–414. LaBella, F.S.; Templeton, J.F. Clin. Exp. Hypertens. 1998, 20, 601–609. [PubMed]
- Kim, R.S.; LaBella, F.S.; Zunza, H.; Zunza, F.; Templeton, J.F. Mol. Pharmacol. 1980, 18, 402–405. [PubMed]LaBella, F.S.; Bihler, I.; Templeton, J.F.; Kim, R.S.; Hnatowich, M.; Rohrer, D.C. Fed. Proc. 1985, 44, 2806–2811. [PubMed]
- Templeton, J.F.; Ling, Y.; Marat, K.; LaBella, F.S. J. Med. Chem. 1997, 40, 1439–1446. [PubMed]Templeton, J.F.; Kumar, V.P.S.; Bose, D.; LaBella, F.S. J. Med. Chem. 1989, 32, 1977–1981. [PubMed]Templeton, J.F.; Ling, Y.; Jin, J.; Boehmer, M.A.; Zeglam, T.H.; LaBella, F.S. J. Chem. Soc. Per-kin Trans. I 1991, 823–829. Templeton, J.F.; Kumar, V.P.S.; Bose, D.; Smyth, D.D.; Kim, R.S.; LaBella, F.S. Can. J. Physio. Pharmacol. 1988, 66, 1420–1424. Smyth, D.D.; Templeton, J.F.; Kumar, V.P.S.; Yan, Y.; Widajewicz, W.; LaBella, F.S. Can. J. Physiol Pharmacol. 1992, 70, 723–727. [PubMed]Templeton, J.F.; Ling, Y.; Zeglam, T.H.; Marat, K.; LaBella, F.S. J. Chem. Soc. Per-kin Trans. I 1992, 2503–2517.
- Templeton, J.F.; Ling, Y.; Zeglam, T.H.; LaBella, F.S. J. Med. Chem. 1993, 36, 42–45. [PubMed]
- Annual Drug Data Report; J.R. Prous Science Publishers: Barcelona, 1995; p. 912.
- Maixent, J.M.; Berrebi-Bertrand, I.; Lelièvre, L.G.; Fenard, S. Arzneim. Forsch. 1992, 42, 1301–1305.
- Swynghedauw, B.; Jarreau, F.X.; Nittemberg, A.; Mouas, C.; Preteseille, M.; Lelièvre, L.G. J. Mol. Cell. Cardiol. 1983, 15 (suppl. 2), 55.
- Repke, K.R.H.; Weiland, J.; Menke, K.H. J. Enzyme Inhib. 1991, 5, 25–32.
- Fullerton, D.S.; Kitatsuji, E.; Deffo, T.; Rohrer, D.C.; Ahmed, K.; From, A.H.L. Curr. Top. Membr. Transp. 1983, 19, 257–264.
- Weiland, J.; Schwabe, K.; Hübler, D.; Schönfeld, W.; Repke, K.R.H. J. Enzyme Inhib. 1987, 2, 31–36. [PubMed]
- Silverstein, M.N.; Petit, R.M.; Solberg, L.M. Am. J. Med. 1992, 92, 69–72. Annual Data Drug Report; J.R. Prous Science Publishers: Barcelona, 1995; p. 914.
- Quadri, L.; Barassi, P.; Gobbini, M.; Fedrizzi, G.; Santagostino, M.; Zappavigna, M.P.; Melloni, P. XVth Symposium on Medicinal Chemistry; Edinburgh (United Kingdom), 1998; Communica-tion P.217. [Google Scholar]
- Szent-Györgyi, A. Chemical Physiology of Contraction in Body Heart Muscle; Academic Press: New York, 1953; pp. 79–88. [Google Scholar]
- Goto, A.; Yamada, N.; Yagi, N.; Yoshioka, M.; Sugimoto, Y. Pharmacol. Rev. 1992, 44, 377–399. [PubMed]Hamlyn, J.M.; Manunta, P. J. Hypertens. 1992, 10, S99–S111. Crambert, G.; Balzan, S.; Paci, A.; Decollogne, S.; Montali, U.; Ghione, S.; Lelièvre, L.G. Ann. N.Y. Acad. Sci. 1997, 834, 621–625. [PubMed]
- Goto, A.; Yamada, K. Curr. Opin. Nephrol. Hypertens. 1998, 7, 189–196. [PubMed]
- Tymiak, A.A.; Norman, J.A.; Bolgar, M.; Didonato, G.C.; Lee, H.; Parker, W.L.; Lo, L.C.; Berova, N.; Nakanishi, K.; Haber, E.; Haupert, G.T., Jr. Proc. Natl. Acad. Sci. USA 1993, 90, 8189–8193. [PubMed]
- Hamlyn, J.M.; Blaustein, M.P.; Bova, S.; Ducharme, D.W.; Harris, D.W.; Mandel, F.; Mathews, W.R.; Ludens, J.H. Proc. Natl. Acad. Sci. USA 1991, 88, 6259–6263. [PubMed]
- De Wardener, H.E. J. Hypertens. 1996, 14, S9–S18.
- Carilli, C.T.; Berne, M.; Cantley, L.C.; Haupert, G.T. J. Biol. Chem. 1985, 260, 1027–1031. [PubMed]
- Haupert, G.T. The Na+,K+ Pump, Part B: Cellular Aspects; Skou, J.C., Nørby, J.G., Mauns-bach, A.B., Esmann, M., Eds.; Alan R. Liss: New York, 1988; pp. 297–320. [Google Scholar]
- Kelly, R.A.; Smith, T.W. Adv. Pharmacol. 1994, 25, 263–288. [PubMed]
- Paci, A.; Sakakibara, M.; Del Bene, P.; Uchida, A.O. Ann. N.Y. Acad. Sci. 1997, 834, 637–641. [PubMed]
- Doris, P.A. Miner. Electrolyte Metab. 1996, 22, 303–310. [PubMed]Hollenberg, N.K.; Graves, S.W. Progress in Drug Research; Birkhäuser Verlag: Basil, 1996; Vol. 46, pp. 9–42. [Google Scholar] Pidgeon, G.B.; Lewis, L.K.; Yandle, T.G.; Richards, A.M.; Nicholls, M.G. J. Hypertens 1996, 14, 169–171.
- Greenhill, J.V.; Lue, P. Progress in Medicinal Chemistry; Ellis, G.P., Luscombe, D.K., Eds.; Elsevier: Amsterdam, 1993; Vol. 30, pp. 203–326. [Google Scholar]
- Greenhill, J.V.; Ismail, M.J.; Edwards, P.N.; Taylor, P.J. J. Chem. Soc. Perkin Trans. II 1985, 1255–1264.
- Bryant, H.U.; Nelson, D.L.; Button, D.; Cole, H.W.; Baez, M.B.; Lucaites, V.L.; Wainscott, D.B.; Whitesitt, C.; Reel, J.; Simon, R.; Koppel, G.A. Life Sci. 1996, 59, 1259–1268. [PubMed]Bubner, M.; Kasbohm, K.; Heise, K.H.; Richter, P.H. Pharmazie 1995, 50, 71–72. [PubMed]Dorhout, B.; Poortenga, P.J.; Kingma, A.W.; De Hoog, E.; Muskiet, F. Biochim. Biophys. Acta 1998, 1381, 95–103. [PubMed]Desideri, N.; Sestili, I.; Piccardoni, P.; Rotondo, S.; Cerletti, C.; Stein, M.L. Arch. Pharm. (Weinheim) 1992, 325, 773–777. [PubMed]Diamant, S.; Agranat, I.; Goldblum, A.; Cohen, S.; Atlas, D. Biochem. Pharmacol. 1985, 34, 491–498. [PubMed]Doubell, P.C.; Oliver, D.W. Arzneim. Forsch. 1992, 42, 65–69. Mukhopadhyay, R.; Kapoor, P.; Madhubala, R. Pharmacol. Res. 1996, 33, 67–70. [PubMed]
- Ng, Y.C.; Leung, W.Y.; Akera, T. Eur. J. Pharmacol. 1988, 155, 93–99. [PubMed]
- Thomas, R.E.; Boutagy, J.; Gelbart, A. J. Pharm. Sci. 1974, 63, 1649–1683. [PubMed]
- Lingrel, J.B.; Kuntzweiler, T. J. Biol. Chem. 1994, 269, 19659–19662. [PubMed]Shull, G.E.; Lane, L.K.; Lingrel, J.B. Nature 1986, 321, 429–431. [PubMed]
- Gelbart, A.; Thomas, R.E. J. Med. Chem. 1978, 21, 284–288. [PubMed]
- Herber, D.; Herzig, S.; Moosig, F.; Neujahr, H. Pharmazie 1995, 50, 663–667.
- Thomas, R.E.; Gray, P.; Andrews, J. Adv. Drug Res. 1990, 19, 814–839.
- Cerri, A.; Serra, F.; Ferrari, P.; Folpini, E.; Padoani, G.; Melloni, P. J. Med. Chem. 1997, 40, 3484–3488. [PubMed]Schutz, S.; Meyer, K.; Kratzer, H. Arzneim. Forsch. 1969, 19, 69–75.
- David, P.; Mayan, H.; Cohen, H.; Tal, D.M.; Karlish, S.J. J. Biol. Chem. 1992, 267, 1141–1149. [PubMed]
- Sample availability: Samples in references 52-55 of this review are availables from authors










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
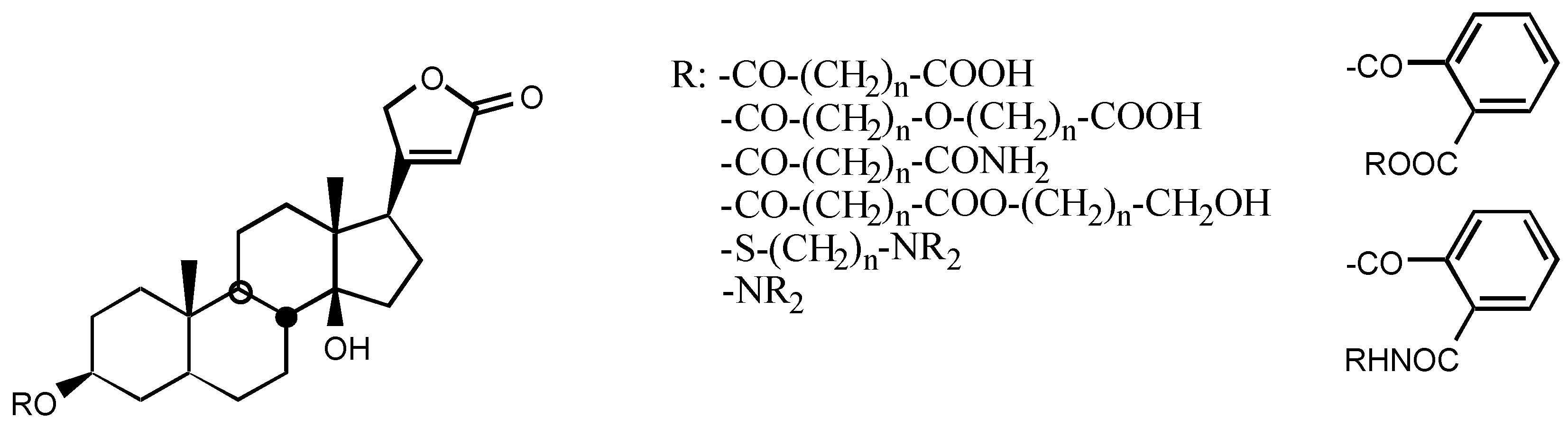{kind=link}
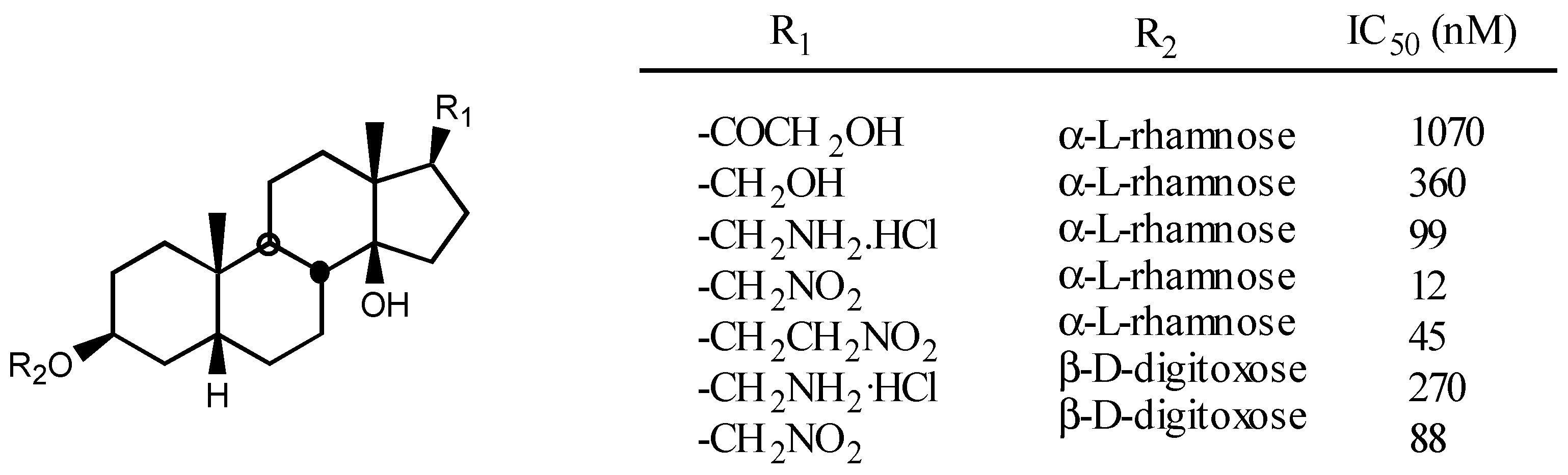{kind=link}
{kind=link}
{kind=link}
{kind=link}
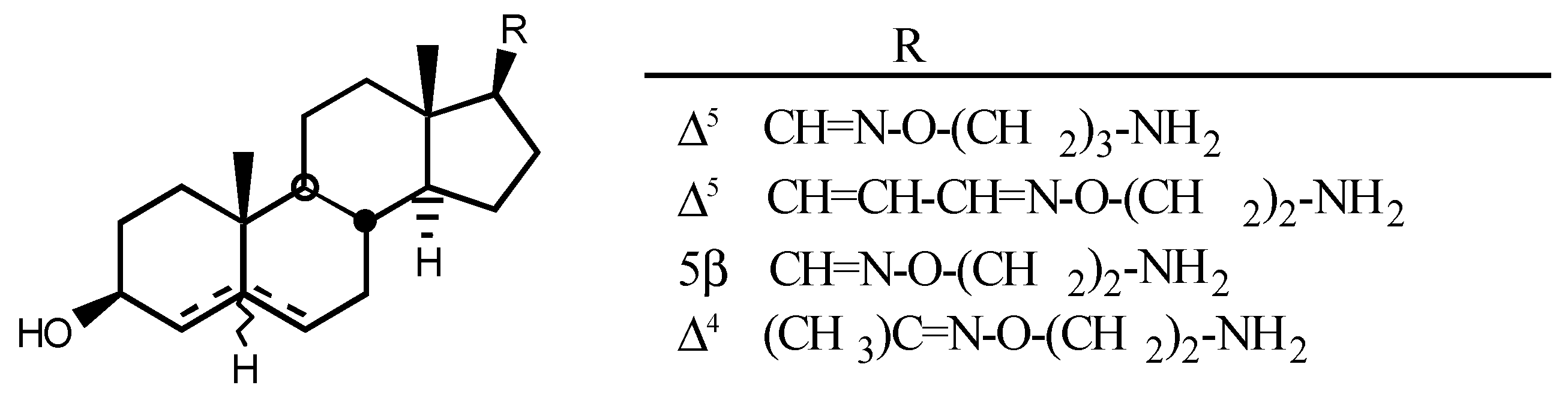{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Cardiotonic glycosides |
|---|---|---|
| Apocynaceae | Nerium oleander | Oleandrin, neriin, neriantin. |
| Nerium odorum | Odoroside A and B. | |
| Strophantus gratus, S. kombe, S. his-pidus, S. sarmentosus, S. emini | Ouabain (G-strophantin), cymarin, sarmentocyma-rin, periplocymarin, K-strophantin. | |
| Acokanthera schimperi (A. ouabaïo), A. venenata, A. abyssinica | Ouabain. | |
| Thevetia nereifolia | Thevetin, cerberin, peruvoside. | |
| Thevetia yecotli | Thevetosin, thevetin A. | |
| Cerbera odollam | Cerberin. | |
| Cerbera tanghin | Tanghinin, deacetyltanghinin, cerberin. | |
| Adenium boehmanianum | Echujin, hongheloside G. | |
| Asclepiadaceae | Periploca graeca | Periplocin. |
| Periploca nigrescens | Strophantidin, strophantidol, nigrescin. | |
| Xysmalobium undulatum | Uzarin. | |
| Gomphocarpus fruticosus | Uzarin. | |
| Calotropis procera | Calotropin. | |
| Brassicaceae | Cheiranthus cheiri | Cheiroside A, cheirotoxin. |
| Celastraceae | Euonymus europaeus, E. atropur-pureus | Eounoside, euobioside, euomonoside. |
| Crassulaceae | Kalanchoe lanceolata | Lancetoxin A and B. |
| Kalanchoe tomentosa | Kalanchoside. | |
| Kalanchoe tubiflorum | Bryotoxin A-C. | |
| Kalanchoe pinnatum | Bryotoxin C, bryophyllin B. | |
| Tylecodon wallichii | Cotiledoside. | |
| Tylecodon grandiflorus | Tyledoside A-D, F and G. | |
| Cotyledon orbiculata | Orbicuside A-C. | |
| Fabaceae | Coronilla sp. | Alloglaucotoxin, corotoxin, coroglaucin, glau-corin. |
| Iridaceae | Homeria glauca | Scillirosidin derivatives. |
| Moraea polystachya, M. graminicola | Bovogenin A derivatives. | |
| Liliaceae | Urginea scilla, U. maritima | Scillarene A and B, scilliroside, scillarenia, scillia-cinoside, scilliglaucoside, scilliglaucosidin, scil-liphaeosidin, scilliphaeoside, scillirosidin, scilliru-brosidin, scillirubroside, proscillaridin A. |
| Urginea rubella | Rubelin. | |
| Convalaria majalis | Convalloside, convallatoxin. | |
| Bowiea volubilis, B. kilimand-scharica | Bovoside A, glucobovoside A, bovoruboside. | |
| Moraceae | Antiaria africana, A. toxicaria | Antiarin α. |
| Ranunculaceae | Helleborus niger, H. viridis, H. foeti-dus | Helleborein, helleborin, hellebrin. |
| Adonis vernalis, A. aestivalis, A.autumnalis, A. flammea | Adonidin, adonin, cymarin, adonitoxin. | |
| Santalaceae | Thesium lineatum | Thesiuside. |
| Scrophulariaceae | Digitalis purpurea, D. lanata | Digitoxin, gitoxin, gitalin, digoxin, F-gitonin, digitonin, lanatoside A-C. |
| Species | Genins |
|---|---|
| Bufo vulgaris | Bufotalin, bufotalinin, bufotalidin. |
| Bufo japonicus | Gamabufagin. |
| Bufo gargarizans | Cinobufagin. |
| Bufo marinus | Marinobufagin. |
| Bufo arenarum | Arenobufagin. |
| Bufo regularis | Regularobufagin. |
| Bufo valliceps | Vallicepobufagin. |
| Bufo quercicus | Quercicobufagin. |
| Bufo viridis | Viridibufagin. |
| Bufo sp. | Pseudobufotalin. |
© 2000 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Melero, C.P.; Medarde, M.; San Feliciano, A. A Short Review on Cardiotonic Steroids and Their Aminoguanidine Analogues. Molecules 2000, 5, 51-81. https://doi.org/10.3390/50100051
Melero CP, Medarde M, San Feliciano A. A Short Review on Cardiotonic Steroids and Their Aminoguanidine Analogues. Molecules. 2000; 5(1):51-81. https://doi.org/10.3390/50100051
Chicago/Turabian StyleMelero, Concepción P., Manuel Medarde, and Arturo San Feliciano. 2000. "A Short Review on Cardiotonic Steroids and Their Aminoguanidine Analogues" Molecules 5, no. 1: 51-81. https://doi.org/10.3390/50100051
APA StyleMelero, C. P., Medarde, M., & San Feliciano, A. (2000). A Short Review on Cardiotonic Steroids and Their Aminoguanidine Analogues. Molecules, 5(1), 51-81. https://doi.org/10.3390/50100051