Introduction
Pyrrole synthesis depends strongly on the nature and relative position of the substituents in the molecule. The methods permitting the synthesis of comparatively simple pyrroles are found to be use-less for more complex structures. This paper describes the synthesis of a series of pyrrole derivatives (1-5).
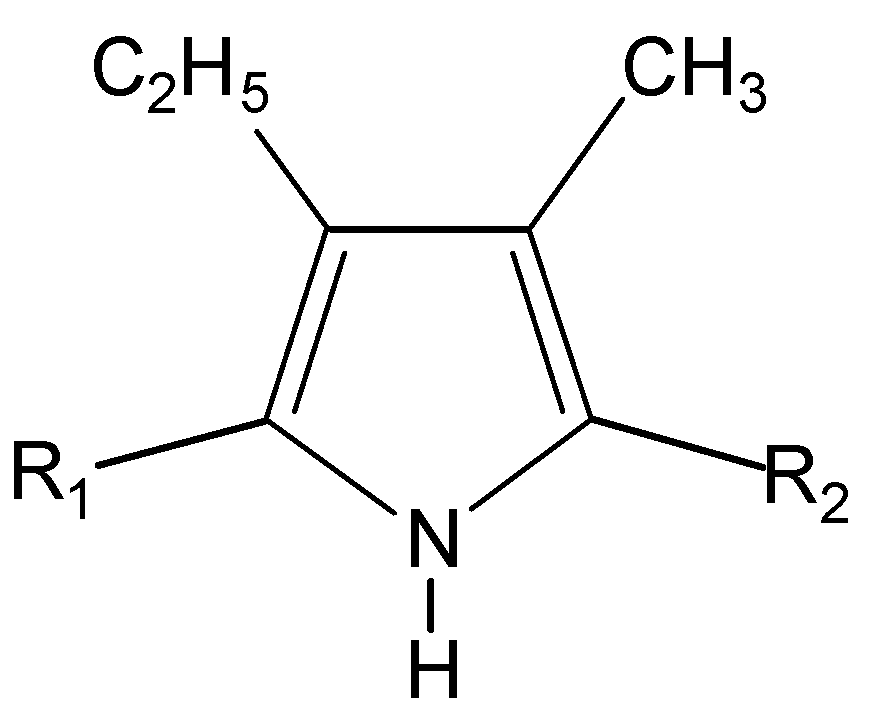
Figure 1.
R1 = CH3 (1, 6), COOC2H5 (2-5); R2 = COOCH2C6H5 (1, 2), COOH (3), I (4), H (5), COOC2H5 (6).
Figure 1.
R1 = CH3 (1, 6), COOC2H5 (2-5); R2 = COOCH2C6H5 (1, 2), COOH (3), I (4), H (5), COOC2H5 (6).
Relatively accessible α-ethoxycarbonylpyrroles are obtained by the condensation of the corre-sponding alkylpentanediones with nitrosoacetoacetic ester in acetic acid with the reduction [
1,
2,
3,
4,
5,
6,
7,
8]. However α-unsubstituted pyrroles – the desired product of the synthesis - have a great disadvantage: they contain large substituents in adjacent β-positions responsible for extra restrictions during conden-sation. We now report a method that allows the synthesis of structures with these large substituents in the desired positions while excluding the steric factor influence on pyrrole reactivity.
Experimental
General Methods and Materials
1H nuclear magnetic resonance spectra were recorded on a Bruker AC-500 spectrometer operating at 600 MHz with HMDS as internal standard in deuterochloroform. IR spectra were obtained on a Spe-cord M80 spectrophotometer in KBr tablets. 2,4-Dimethyl-3-ethyl-5-ethoxycarbonylpyrrole (
6) was synthesized by a known method [
2].
2,4-Dimethyl-3-ethyl-5-benzyloxycarbonylpyrrole (1)
2,4-Dimethyl-3-ethyl-5-ethoxycarbonylpyrrole (6, 10 g) in benzyl alcohol (50 mL) were placed into a flask equipped with a distilling column, Liebig condenser and dropping funnel. The solution was heated to boiling temperature and then a solution of sodium (0.2 g) in benzyl alcohol (100 mL) was added to the flask. After all the ethanol was distilled off the reaction mixture was cooled, diluted with water (200 mL) and then acidified with acetic acid. The resulting precipitate was isolated by filtration and recrystallized from ethanol. Yield 87%, melting point 166-168°C, NMR: 8.89 (s, 1H, NH), 7.34 (m, 5H, C6H5), 4.22 (m, 2H, OCH2Ph), 2.27 (q, 2H, CH2CH3), 2.21 (s, 3H, 2-CH3), 2.09 (s, 3H, 4-CH3), 0.84 (t, 3H, CH2CH3)
2-Ethoxycarbonyl-3-ethyl-4-methyl-5-benzyloxycarbonylpyrrole (2)
Freshly distilled sulfuryl chloride (8.9 mL) was added to a solution of 2,4-dimethyl-3-ethyl-5-benzyloxycarbonylpyrrol (1, 9g) in dry ether (100 mL) while the reaction mixture temperature was kept below 20°C. The mixture was kept overnight at room temperature and then the ether was evapo-rated. Ethanol (150 mL) was added to the residuum. After 1 hour the solution was diluted with water (200 mL). The organic phase was extracted with ether and washed with water. The ether was evapo-rated and the resulting product was recrystallized from ethanol. Yield 92%, melting point 51-53°C, NMR: 8.88 (s, 1H, NH), 7.36 (m, 5H, C6H5), 4.32 (m, 2H, OCH2Ph), 4.13 (q, 2H, OCH2CH3), 2.69 (q, 2H, CH2CH3), 2.24 (s, 3H, 4-CH3), 1.27 (t, 3H, OCH2CH3), 0.84 (t, 3H, CH2CH3).
2-Ethoxycarbonyl-3-ethyl-4-methyl-5-carboxypyrrole (3)
Hydrogen was passed through a stirred mixture of 2-ethoxycarbonyl-3-ethyl-4-methyl-5-benzyloxycarbonylpyrrole (2, 10 g), activated 10% Pd/C (2g), tetrahydrofuran (200 mL) and trieth-ylamine (2 mL) over 6 hours. Then the solution was evaporated down to 50-mL residual volume and methanol (100 mL) was added. After evaporating the remaining tetrahydrofuran the methanol solution was cooled. The resulting precipitate was isolated by filtration and washed with methanol, then ether and hexane. Yield 96%, melting point 186-188°C, NMR: 9.38 (s, 1H, NH), 4.17 (q, 2H, OCH2CH3 ), 2.69 (q, 2H, CH2CH3), 2.24 (s, 3H, 4-CH3), 1.32 (t, 3H, OCH2CH3), 0.87 (t, 3H, CH2CH3).
2-Ethoxycarbonyl-3-ethyl-4-methyl-5-iodopyrrole (4)
A solution of sodium bicarbonate (4.5 g) in water (80 mL) was added dropwise to a stirred solution of 2-ethoxycarbonyl-3-ethyl-4-methyl-5-carboxypyrrole (3, 5g) in ethanol (100 mL). Then the solution was filtered and a solution of iodine (4.6 g) and KI (5.9 g) in water (50 mL) was added to the filtrate with stirring at 60°C. Stirring was continued for another 10 minutes and then the reaction mixture was diluted with water (100 mL). The resulting precipitate was isolated by filtration. Yield 86%, melting point 119-120°C, NMR: 8.90 (s, 1H, NH), 4.21 (q, 2H, OCH2CH3), 2.67 (q, 2H, CH2CH3), 1.90 (s, 3H, 4-CH3), 1.28 (t, 3H, OCH2CH3), 0.86 (t, 3H, CH2CH3).
2-Ethoxycarbonyl-3-ethyl-4-methylpyrrole (5)
Hydrochloric acid (100 mL) was added to a boiling solution of 2-ethoxycarbonyl-3-ethyl-4-methyl-5-iodopyrrole (4, 5g), SnCl2 (8.5 g) and KI (0.8 g) in ethanol (350 mL). The organic phase was ex-tracted with ether and washed with water. The ether was evaporated under vacuum at room tempera-ture. Yield 87%, melting point 22-24°C, NMR: 8.94 (s, 1H, NH), 4.20 (q, 2H, OCH2CH3), 2.68 (q, 2H, CH2CH3), 1.94 (s, 3H, 4-CH3), 1.29 (t, 3H, OCH2CH3), 0.87 (t, 3H, CH2CH3)



{kind=link}