Synthetic procedures and spectral data
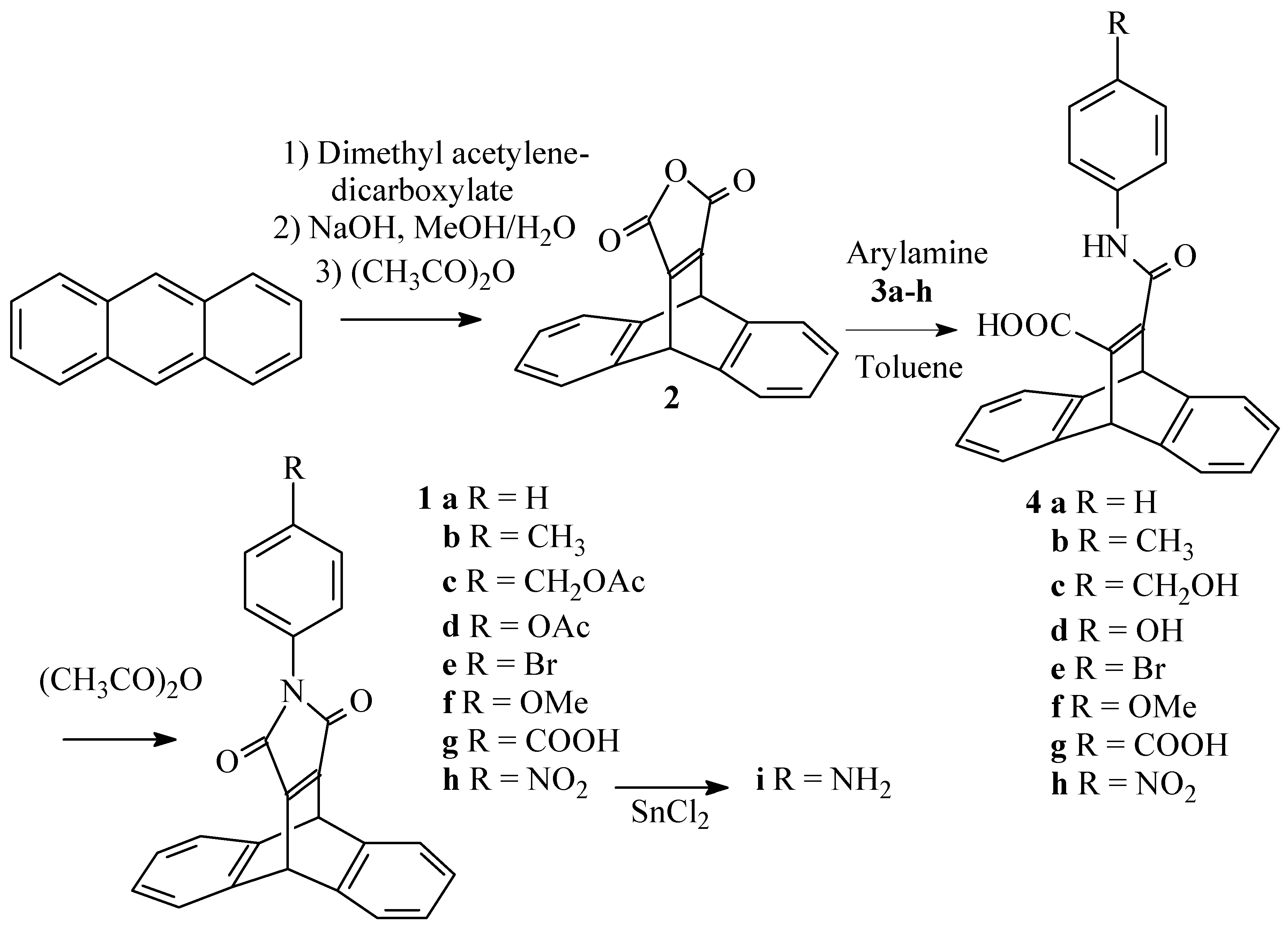
9,10-Dihydro-9,10-ethenoanthracene-11,12-dicarboxylic anhydride (2) [5]
A suspension of anthracene (15 g; 0.084 mol) and dimethyl acetylenedicarboxylate (15 ml) was heated for 45 minutes at 170°C, after which heating is continued for another 5 minutes at 180°C. The reaction mixture is cooled and the adduct crystallized from methanol. This compound was taken up in a solution of 5g of NaOH in a mixture of 75 ml of methanol and 25 ml of water, and the resulting suspension was refluxed for 1 hour. The reaction mixture was cooled down and left overnight at -20°C. The crystals formed were collected by filtration and concentrated hydrochloric acid was added to this salt to liberate the free acid. This was collected by filtration and taken up in warm acetic anhydride (50 ml) in the presence of sodium acetate (0.5 g). The resulting suspension was heated at 80°C for 30 minutes. The reaction mixture as cooled down and the solvent was evaporated in vacuo. The resulting solid was crystallized from ether/hexane (1/2) (60 ml) and obtained in 71% yield (overall): mp : 256°C (lit.4 247°C) ; IR (KBr, cm-1) : 1767 (s br, CO), 1635 (s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) : 5.54 (s, 2H, 9,10-H), 7.00-7.10 (m, 4H, 2,3,6,7-H), 7.36-7.48 (m, 4H, 1,4,5,8-H); 13C-NMR (62.5 MHz/CDCl3) δ (ppm) :47.8,124.9,126.2,142.9,160.4; MS (EI, m/z): 274 M+.
General procedure for the preparation of the maleimides (1a-h)
A suspension of the anhydride (2) (3.62 mmol) and the amine (3a-h) (3.80 mmol) in toluene (10 ml) was refluxed for 30 minutes. The resulting suspension was evaporated in vacuo and acetic anhydride (5 ml) and sodium acetate (0.1 g) were added. The mixture was heated for another 30 minutes at 80°C. The resulting clear solution was again evaporated in vacuo and the compound crystallized from methanol. For compound 1g, the thick slurry, obtained after evaporation of the acetic anhydride is first treated with a few drops of water and stirred for 5h, to destroy the formed mixed anhydride).
N-Phenyl-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1a)
It was obtained following the general procedure in 87% yield: mp: 245°C; IR (KBr, cm-1) : 1768 (s,CO), 1705 (s br, CO), 1630 (s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) :5.61 (s, 2H, 9,10-H), 7.01-7.10 (m, 4H,2,3,6,7-H), 7.19-7.25 (m,2H, m-H), 7.28-7.33 (m, 1H, p-H), 7.36-7.42 (m, 2H, o-H), 7.42-7.49(m, 4H, 1,4,5,8-H); ); 13C-NMR (62.5 MHz/CDCl3) δ (ppm): 47.2, 124.6, 125.6, 126.5, 127.6, 128.9, 131.7, 144.0, 156.2, 165.3; MS (EI, m/z): 349 M+.
N-(4-Methylphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1b)
It was obtained following the general procedure in 94% yield: mp: 250°C; IR (KBr, cm-1) :1766 (s,C=O), 1699 (s br, C=O); 1H-NMR (400 MHz/CDCl3) δ (ppm) :2.34 (s, 3H, CH3), 5.61 (s, 2H, 9,10- H) 7.05-7.07 (m, 4H, 2,3,6,7-H), 7.09 (d, J=8.5Hz, 2H, CHCH3), 7.20 (2H, J=8.5Hz, 2H, CHCHCH3), 7.43-7.45 (m, 4H, 1,4,5,8-H); 13C-NMR (100 MHz/CDCl3) δ (ppm): 21.1, 47.23, 124.6, 125.6, 126.5, 129.0, 129.7, 137.8, 144.0, 156.17, 165.5; MS (EI, m/z): 363 M+. EA(%C,%H,%N): Calcd.: 82.63, 4.71, 3.85, Found: 82.9, 5.0, 3.5.
N-(4-(Acetoxymethyl)phenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1c)
It was obtained following the general procedure in 92% yield: mp: 68°C; IR (KBr, cm-1) : 1714 (s, C=O), 1638 (m,C=C); 1H-NMR (400 MHz/CDCl3) δ (ppm) : 2.09 (s, 3H, CH3), 5.09 (s, 2H, CH2), 5.61, (s, 2H, 9,10-H), 7.05-7.10 (m, 4H, 2,3,6,7-H), 7.24 (d, J=8.5Hz, 2H, CHCCH2), 7.40 (d, 2H, J=8.5Hz, CHN), 7.42-7.48 (m, 4H, 1,4,5,8-H), 13C-NMR (100 MHz/CDCl3) δ (ppm): 20.9, 47.6, 65.6, 124.6, 125.7, 126.5, 128.9, 131.6, 135.3, 143.9, 156.3, 165.2, 170.8; MS (EI, m/z): 421 M+, EA(%C,%H,%N): Calcd.: 76.95, 4.54, 3.32, Found: 77.2, 4.8, 3.1.
N-(4-Acetoxyphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1d)
It was obtained following the general procedure in 83% yield: mp: 296°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) :2.29 (s, 3H CH3) 5.60 (s, 2H, 9,10-H), 7.02-7.09 (m 4H, 2,3,6,7-H), 7.12 (d, J=8Hz, 2H, CHCO), 7.26 (d, J= 8Hz, CHCN), 7.41-7.48 (m, 4H, 1,4,5,8-H); MS (EI, m/z): 407 M+ EA(%C,%H,%N): Calcd.: 76.65, 4.21, 3.44, Found: 76.5, 4.3, 3.5.
N-(4-Bromophenyl)-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboximide (1e)
It was obtained following the general procedure in 90% yield: mp: 276°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) : 5.59 (s, 2H, 9,10-H), 7.04-7.06 (m, 4H, 2,3,6,7-H), 7.12 (d, J=8Hz, 2H, CHCBr), 7.42-7.45 (m, 4H, 1,4,5,8-H), 7.51 (d, J=8Hz, 2H CHCN); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.3, 121.3, 124.6, 125.7, 127.8, 130.8, 132.1, 143.9, 156.4, 164.9; MS (EI, m/z): 427/429 M+ EA(%C,%H,%N, %Br): Calcd.: 67.31, 3.29, 3.27, 18.66, Found: 67.3, 3.3, 3.0, 18.4.
N-(4-Methoxyphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1f)
It was obtained following the general procedure in 90% yield: mp: 301°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) :3.79 (s, 3H, OCH3), 5.60 (s, 2H, 9,10-H), 6.92 (d, J= 8.5Hz, 2H, CHCOCH3), 7.04-7.08 (m, 4H, 2,3,6,7-H), 7.12 (d, J=8.5Hz, 2H, CHCN), 7.42-7.46 (m, 4H, 1,4,5,8-H); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.3, 55.4, 114.4, 124.3, 125.7, 128.1, 144.1, 156.2, 159.0, 165.7; MS (EI, m/z): 379 M+ EA(%C,%H,%N): Calcd.: 79.14, 4.52, 3.69, Found: 79.1, 4.9, 3.2.
N-(4-Carboxyphenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1g)
It was obtained following the general procedure in 90% yield: mp: 307°C; 1H-NMR (400 MHz/DMSO-d6) δ (ppm) :5.84 (s, 2H, 9,10-H), 7.05-7.07 (m, 4H, 2,3,6,7-H), 7.45 (d, J=7.5Hz, 2H, CHCN), 7.56-7.58 (m, 4H, 1,4,5,8-H), 7.98 (d, J=7.5Hz, 2H, CHCCOOH), 12-13 (s br, 1H COOH); 13C-NMR (100 MHz/DMSO-d6) δ (ppm): 46.1, 124.6, 125.3, 126.5, 129.4, 129.7, 135.8, 144.3, 155.8, 164.5, 166.7; MS (EI, m/z): 393 M+.
N-(4-Nitrophenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1h)
It was obtained following the general procedure in 95% yield: mp: 311°C; 1H-NMR (400 MHz/CDCl3) δ (ppm) : 5.62 (s, 2H, 9,10-H), 7.07-7.09 (m, 4H, 2,3,6,7-H), 7.45-7.48 (m, 4H, 1,4,5,8- H), 7.55 (d, J = 7.5 Hz, 2H, CHCN), 8.27 (d, J = 7.5 Hz, 2H, CHCNO2); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.33, 124.4, 124.7, 125.7, 125.9, 137.8, 143.6, 145.9, 156.8, 164.4; MS (EI, m/z): 394 M+.
N-(4-Aminophenyl)-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboximide (1i)
A solution of 1h (0.3g; 0.77mmol) and anhydrous SnCl2 (4g) in THF (8ml) was stirred for 20 h. The mixture was poured on crushed ice (20g) and the resulting slurry was extracted with dichloromethane (3x30ml). The combined organic layers were dried over MgSO4 and evaporated in vacuo. The compound was obtained as a thick oil, after column chromatography (silica) with dichloromethane as the eluent, in 86% yield. 1H-NMR (250 MHz/CDCl3) δ (ppm) : 3.3-3.8 (sbr, 2H, NH2), 5.59 (s, 2H, 9,10-H), 6.63 (d, J=8Hz, 2H, CHCNH2), 6.92 (d, J=8Hz, 2H, CHCN), 7.02-7.08 (m, 4H, 2,3,6,7-H), 7.48-7.55 (m, 4H, 1,4,5,8-H); MS (EI, m/z): 364 M+.
General procedure for the preparation of the maleimides 7a,b and 8
A supension of the anhydride (2) (3.62 mmol) and the appropriate diamine (17mmol) in toluene (10 ml) was refluxed for 30 minutes. The resulting suspension was evaporated in vacuo and acetic anhydride (5 ml) and sodium acetate (0.1 g) were added. The mixture was heated for another 30 minutes at 80°C. The resulting clear solution was again evaporated in vacuo and the compound crystallized from methanol. For compound 7b, the thick slurry, obtained after evaporation of the acetic anhydride is first treated with a few drops of water and stirred for 5h, to hydrolyze the formed mixed anhydride.
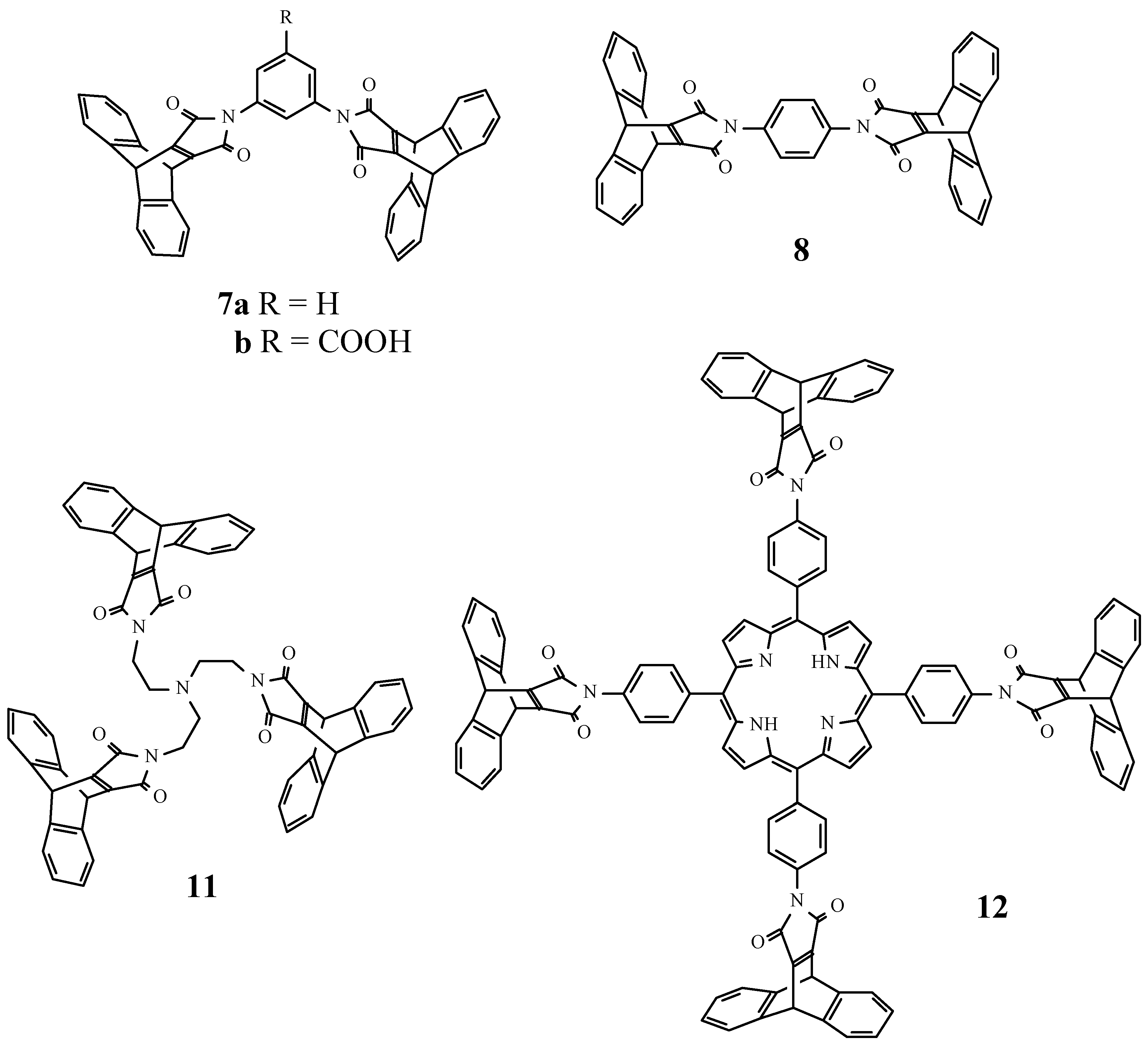
Bismaleimide (7a)
It was obtained following the general procedure in 83% yield: mp: >360°C; IR (KBr, cm-1) : 1773 (s, C=O), 1714 (s, C=O), 1630 (s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) : 5.56 (s, 4H, 9,10-H), 7.00-7.07 (m, 8H, 2,3,6,7-H), 7.19-7.24 (m, 3H, 1,3-disubstituted phenyl-H), 7.36-7.44 (m, 9H, 1,4,5,8-H and NCHN); 13C-NMR (62.5 MHz/CDCl3) δ (ppm): 47.2, 123.4, 124.5, 124.7, 125.7, 129.1, 132.4, 143.9, 156.3, 164.8; MS (EI, m/z): 621 MH+, EA(%C,%H,%N): Calcd.: 81.28, 3.90, 4.51, Found: 81.7, 3.9, 4.1.
Bismaleimide (7b)
It was obtained following the general procedure in 81% yield: mp: >360°C; 1H-NMR (250MHz/CDCl3) δ (ppm) : 5.58 (s, 4H, 9,10-H), 6.98-7.02 (m, 8H, 2,3,5,6-H), 7.38-7.42 (m, 8H, 1,4,5,8-H), 7.48 (t, J=1Hz, 1H, 4-H phenyl), 7.93 (d, J=1Hz, 2H, 2,6-H phenyl); 13C-NMR (62.5MHz/CDCl3) δ (ppm): 47.27, 124.63, 125.76, 126.06, 127.94, 130.77, 132.72, 143.75, 156.46, 164.50, 169.31; MS (EI, m/z): 665 M+.
Bismaleimide (8)
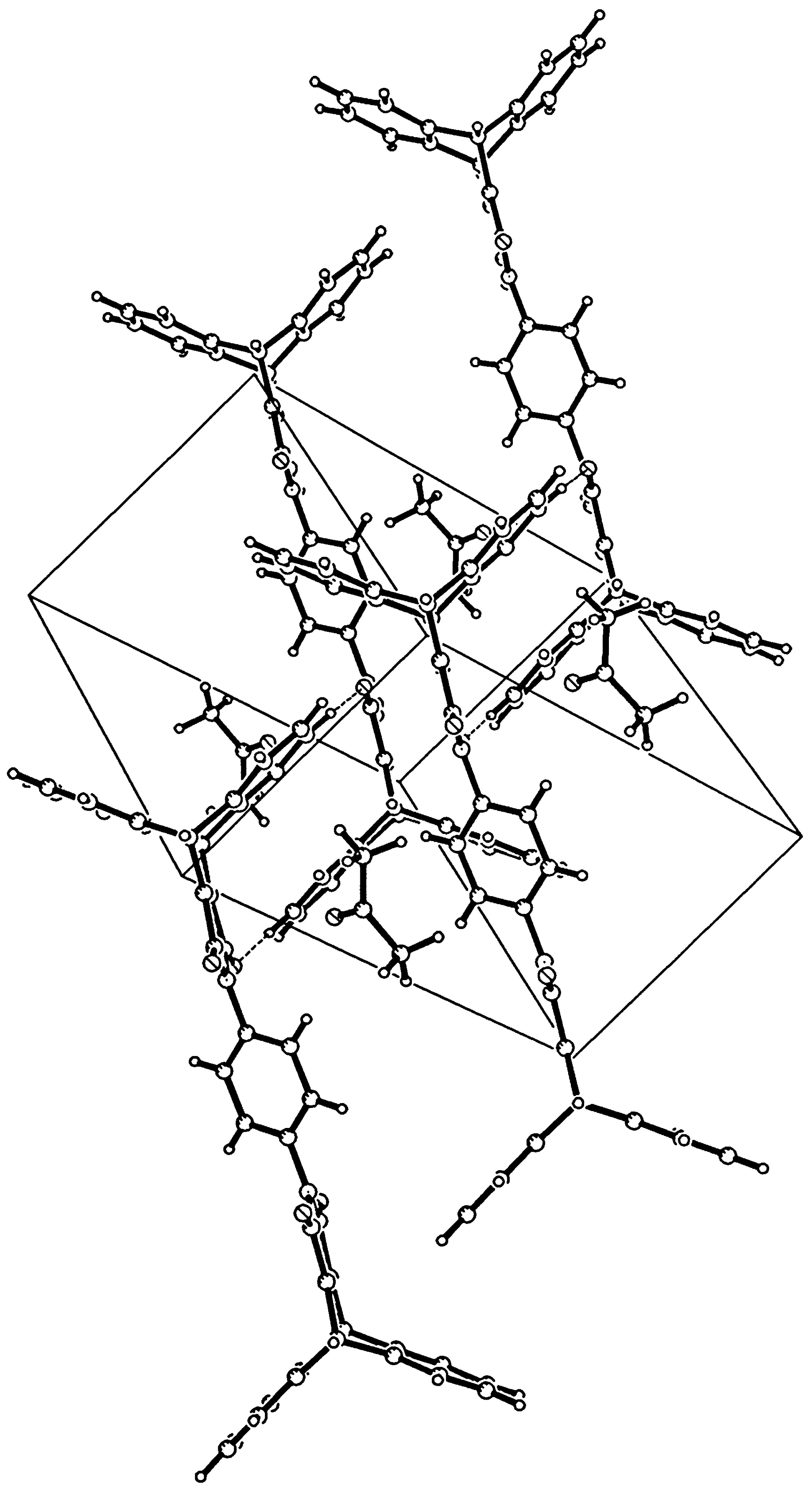
It was obtained following the general procedure in 93% yield: mp: >360°C; IR (KBr, cm-1) : 1769 (s, C=O), 1709 (s br, C=O, s, C=C); 1H-NMR (250 MHz/CDCl3) δ (ppm) : 5.52 (s, 4H, 9,10-H), 7.06- 7.14 (m, 8H, 2,3,5,6-H), 7.35 (s, 4H, 1,4-disubstituted phenyl-H), 7.46-7.53(m, 8H, 1,4,5,8-H); 13C-NMR (62.5 MHz/CDCl3) δ (ppm): 47.3, 124.6, 125.7, 126.7, 130.9, 143.9, 156.3, 165.0; MS (EI, m/z): 620 M+; The compound was recrystallized from acetone for X-ray analysis. Crystal data (acetone) : C42H24N204.2C3H60, Mr = 736.79, light yellow plates (0.40 x 0.35 x 0.04 mm), triclinic, space group P- 1 (no. 2) with a = 8.1320(10), b = 10.742(2), c = 12.175(2) Å, α = 106.92(1), β = 105.11(1), γ = 98.65(1)°, V = 952.4(3) Å3, Z = 1, Dc = 1.285g cm-3, F(000) = 386, μ(Mo-Kα) = 0.085 mm-1, 4012 reflections measured, 3253 independent, Rint = 0.0344, 1.8° < θ < 24.7°, ω scan, T = 289K, Mo-Kα radiation, graphite monochromator, λ = 0.71073 Å on a Siemens P4 diffractometer. Data were corrected for Lp effects, but not for absorption. The structure was solved by direct methods (SHELXTL). Refinement on F2 was carried out by full-matrix least-squares techniques (SHELXTL) with anisotropic displacement parameters for non-H atoms and riding isotropic H-atoms. Refinement converged at a final wR2 value of 0.1217, R1 = 0.0471 for 2161 reflections with Fo > 4σ(Fo), S = 10016, 269 parameters. A final difference Fourier showed no residual density outside -0.20 and 0.15 e Å-3.
Trismaleimide 11
A solution of the anhydride (2) (500mg; 1.8 mmol) and tris(2-aminoethyl)amine (66mg; 0.45 mmol) in dry THF (30 ml) was stirred for 3 h. The solvent was evaporated in vacuo and acetic anhydride (10 ml) and sodium acetate (0.1 g) were added and the mixture heated for 30 minutes at 80°C. The solvent was evaporated and the compound was obtained as a viscous oil, after purification by column chromatography (silica) with dichloromethane/diethyl ether (2:1) as the eluent, in a 61% yield: IR (CCl4, cm-1): 1707 (s, C=O), 1636 (m, C=C); 1H-NMR (400 MHz/CDCl3) δ (ppm) : 2.46 (t, J=6Hz, 6H, CH2N), 3.12 (t, J=6Hz, 6H, CH2N(CO)2), 5.45 (s, 6H, 9,10-H), 6.87-6.91 (m, 12H, 2,3,5,6-H), 7.32-7.36 (m, 12H, 1,4,5,8-H); 13C-NMR (100 MHz/CDCl3) δ (ppm): 36.1, 47.1, 52.2, 124.4, 125.4, 144.1, 156.2, 166.1; (EI, m/z): 914 M+.
Tetrakismaleimide 12
A suspension of the anhydride (2) (220mg; 0.8mmol) and 5,10,15,20-tetrakis(4-aminophenyl)- porphyrin (80mg; 0.118mmol) was refluxed in toluene (40 ml) for 12 h. The solvent was evaporated and acetic anhydride (20ml) and sodium acetate (0.1mg) were added and the mixture heated for 2 h at 80°C. The solvent was stripped off in vacuo and the compound 12 was obtained in 59% yield after purification by column chromatography (silica) with dichloromethane as the eluent: mp >360°C; IR (KBr, cm-1) : 3500 (w; NH), 3370 (w, NH), 1723 (s, C=O), 1638 (m, C=C); 1H-NMR (400 MHz/CDCl3) δ (ppm) : -2.86 (sbr, 2H, NH), 5.80 (s, 8H, 9,10-H (anthracene)), 7.07-7.19 (m, 16H, 2,3,6,7-H), 7.62 (d, J=8Hz, 8H, 3,5-H (phenyl)), 8.20 (d, J=8Hz, 8H, 2,6-H (phenyl)), 8.82 (s, 8H, 2,3,7,8,12,13,17,18-H (porphyrin)); 13C-NMR (100 MHz/CDCl3) δ (ppm): 47.4, 119.3, 124.3, 124.7, 125.8, 131.6, 135.0, 141.1, 144.0, 156.5, 165.5.

{kind=link}
{kind=link}
{kind=link}


