Introduction
The reaction of triorganostannyl ions as nucleophiles with haloarenes has long been known, and the prod- ucts obtained depend on the nucleophile, solvent, and on the reaction conditions. Thus, the reactions of so- dium trimethyltin (NaSnMe
3) with halobenzenes (chloro
-, bromo
- and iodo
-) in tetraglyme as solvent afford phenyltrimethyltin (Me
3SnPh) and variable amounts of reduction product benzene along with diphenyldimeth- yltin (Ph
2SnMe
2) and tetramethyltin (SnMe
4). From trapping experiments and solvent effects it has been pro- posed that the reaction occurs by a halogen metal exchange (HME) mechanism in a solvent cage. The forma- tion of secondary products, Ph
2SnMe
2 and SnMe
4, has been ascribed to the decomposition of NaSnMe
3 into NaMe and dimethylstannylene [
1]. The reaction of
o-dibromobenzene with NaSnMe
3 affords the disubstitu- tion product
o-bis(trimethylstannyl) benzene in 42% yield. In this reaction, the intermediate
o-bromophenyl anion, can decompose into benzyne, which can then be trapped with furan to render the corresponding Diels-Alder adduct [
2].
The reaction of
o-,
m- and
p-bromotoluenes with LiSnBu
3 in THF as solvent affords the expected substitu- tion product, but when
p-chloro and
p-fluorotoluenes are used as substrates,
cine substitution products are obtained, indicating that a benzyne mechanism operates. When radical traps are added, cine substitution prod- ucts increase in yields, and in the presence of Li metal, the yields of
ipso products are enhanced. According to these results, the reaction should proceed, at least in part, by a radical mechanism [
3]. No light stimulation was employed in these reactions.
There are several methods of synthesis for trialkylarylstannanes, typically by the reaction of aryl lithium or organomagnesium derivatives with trialkyltin halides. These reactions have the drawback that many substituents on the aromatic ring are incompatible with the formation of aryl lithium or organo
-magnesium derivatives [
4]. We have described the photostimulated reactions of haloarenes with triorganylstannyl ions by the S
RN1 mechanism. These reactions afford good to excellent yields of the nucleophilic substitution products. Many substituents are compatible with the S
RN1 mechanism, such as CO
2-, CO
2R, CONR
2, RO
-, -CN, R, aryl, NH
2, NR
2 and SO
2R. [
5] Another approach is the palladium catalysis (Stille reaction) of aryl halides [
6] or aryl triflates [
7] with hexamethyl- and hexabutyl-distannanes. Bis(trimethylstannyl) arenes can also be synthesized by palladium catalysis; the yields of disubstitution products range from 40 to 60% [
8]. There are few examples involving reactions of bis(trimethylstannyl) arenes and heteroarenes with aryl halides, which afford a double arylation by the palladium cross-coupling reaction. The examples known afford modest to good yields (6-85% yield) of double arylation [
9,
10,
11].
The S
RN1 mechanism is a chain process, whose main steps are presented in
Scheme 1.
Overall, eqs. 1-3 depict a nucleophilic substitution (eq. 1,3) in which radicals and radical anions are inter- mediates. This chain process requires an initiation step. In a few systems, spontaneous electron transfer (ET) from the nucleophile to the substrate has been observed. When the ET does not occur spontaneously, it can often be induced by light stimulation.
In this review we present the results of the reactions of organotin nucleophiles with aryl halides by the S
RN1 mechanism and their synthetic applications. Reactions are compiled in
Table 1 and
Table 2.
2. Me3Sn- Ions as Nucleophile
Trimethylstannyl anion (Me
3Sn
-) is prepared in liquid ammonia through the reaction of Me
3SnCl with Na metal.
p-Chloroanisole (
1) does not react with Me
3Sn
- ions in the dark after 1 h, but upon irradiation (Hg lamp, 366 nm, 1 h), substitution product
2 was obtained in ca. 100% yield (Table I) (eq. 4). The photostimulated reaction is inhibited by
p-dinitrobenzene, a well-known inhibitor of S
RN1 processes [
12].
Conversely, p-bromoanisole in the presence of Me3Sn- ions affords exclusively anisole in the dark. This reaction proceeds by a HME, with a very fast protonation of the p-anisyl anion by liquid ammonia, anisole being the only product obtained. Bromides and iodides will likely react by a HME reaction faster than by the SRN1 mechanism. When 2-chloroquinoline is allowed to react in the dark with Me3Sn- ions, low yields of the respectively-substituted product are obtained, a reaction which is inhibited by p-DNB and accelerated by light (96%). Other examples are indicated in Table I.
Aryldiethylphosphates are as good leaving groups as chlorines, and they both react with amide ions, NH
2-, to afford substitution products by the S
RN1 mechanism [
13]. The synthesis of aryl trimethylstannanes from phenols through aryldiethylphosphates has recently been reported to proceed in high yields in liquid ammonia. For instance, the photostimulated reaction of
3 with Me
3Sn
- ions affords the substitution product
4 in 93% yield (eq. 5) [
14]. These reactions do not occur in the dark.
Phenyltrimethyl ammonium salts also react with Me
3Sn
- ions in liquid ammonia to give the substitution prod- uct in high yields (Table I) [
15].
When the aromatic substrate bears two leaving groups, such as p-dichlorobenzene (5), disubstitution prod- uct 6 is isolated in 88% yield (eq. 6). The monosubstitution product is not an intermediate in these reactions.
With two different leaving groups, such as in 7, compound 6 is obtained in excellent yields as well (eq. 7).
Substrates such as 2,5-, 2,6- and 3,5- dichloropyridines afford the respective disubstitution products in 80-86% yields [
16] (Table I). There is a report where several chloro, bromo, dichloro and dibromo pyridines and quinolines react with Me
3Sn
- ions in dimethoxyethane to render the mono and disubstitution products in 60-88% yields. There is no photostimulation in these reactions, and no information is provided about the mechanism [
17].
We have found that the photostimulated reaction of 1,3,5-trichlorobenzene 8 in the presence of an excess of Me3Sn- ions affords 71% of the trisubstitution product 9 (eq. 8).
3. Ph3Sn- Ions as Nucleophile
Triphenylstannyl ion (Ph
3Sn
-) was prepared either from reaction of Ph
3SnCl or Ph
3SnSnPh
3, with Na metal in liquid ammonia.
p-Chloro or
p-bromotoluenes
10 do not react with Ph
3Sn
- ions in the dark in liquid ammo- nia as solvent, but upon irradiation, substitution product
11 is obtained in good yields (eq. 9) (
Table 2).
p- Chloroanisole, under irradiation in DMSO, affords only 30% yield of the substitution product.
However, when
p-iodotoluene and
p-iodoanisole are utilized as substrates, a fast HME reaction takes place, affording the reduced product in the dark. Upon irradiation, low yields of substitution products are ob- tained by the S
RN1 mechanism, which competes with the HME reaction (
Table 2). In DMSO as solvent, only the HME reaction is observed.
1-Chloro and 1-bromo naphthalenes afford good yields of the substitution product when irradiated in liquid ammonia in the presence of Ph3Sn- ions. In DMSO as solvent, 1-chloronaphthalene affords good yields of substitution, i.e.: 1-(triphenylstannyl)naphthalene, however, the bromo derivative gives only the HME reaction. From these results it is concluded that the bromo derivatives react mainly by HME reaction in DMSO, but good yields of substitution products are obtained in liquid ammonia. 1- and 2- Naphthyldiethyl-phosphates 12 render excellent yields of substitution products 13 when irradiated in liquid ammonia in the presence of Ph3Sn- ions (eq. 10).
![Molecules 05 01068 i007]()
When p-dichlorobenzene is allowed to react with Ph3Sn- ions under photostimulation in liquid ammonia or DMSO, good yields of the disubstitution product are obtained (60% or 90% respectively.) When p- dibromobenzene is utilized as substrate, there is a fast HME reaction to afford PhBr. Upon irradiation, the disubstitution product, i.e.: p-bis(triphenyltin) benzene, is obtained in only 22% yield, along with tetraphenyltin, which arises from reaction of PhBr with Ph3Sn- ion.
p-Dibromobenzene reacts with Ph
3Sn
- ions in the absence of photostimulation to afford 96% yield of PhBr. In the case of ArI as substrates and Ph
3Sn
- ions as nucleophile, the predominant reaction is the HME in liquid ammonia (
Table 2). However, substrate
14, affords excellent yields of the disubstitution product
15 (eq. 11).
Heterocycles afford high yields of substitution products when the irradiations are conducted in the presence of Ph
3Sn
- ions in liquid ammonia or DMSO. Thus, in DMSO, 2- and 3-chloropyridines afford, the substitution products in 82% and 93% yields respectively; and in liquid ammonia, 2-chloroquinoline furnishes 2- (triphenyltin)quinoline in 80% yield (
Table 2).
4. Other Tin-derived Nucleophiles
When
p-anisyltrimethyltin
2 reacts with sodium metal in liquid ammonia, the Sn-Me bond is cleaved to ren- der nucleophile
16, after neutralization of the generated amide ions with t-butyl alcohol (eq. 12) [
19]. This re- sult is consistent with the bond dissociation energy difference between the tin-phenyl bond (347 kJ mol
-1) [
20] and the tin-methyl bond (259-272 kJ mol
-1) [
20,
21]. Nucleophile
16 was then allowed to react with
p- chlorotoluene
17 under irradiation to afford substitution product
18 in almost quantitative yields (eq. 13).
Product 18 was also obtained in a one-pot fashion starting from Me3Sn- and p-chloroanisole under pho- tostimulation. Intermediate product 2 was not isolated, but treated in situ with Na metal to afford nucleophile 16, which by a subsequent photostimulated reaction in the presence of 17 rendered product 18 in 89% overall yield.
The photostimulated reaction of
p-dichlorobenzene
5 with Me
3Sn
- ions in liquid ammonia affords the disub- stitution product (
6) (eq. 6). When this product is treated
in situ first with Na metal and then with t-BuOH, dianion
19 is obtained, which upon addition of PhCl and ulterior irradiation (90 min) affords product
20 in a one-pot process (70%) [
16] (eq. 14).
When 18 is treated with Na metal in liquid ammonia under the same reaction conditions as those employed above, nucleophile 21 is formed. Addition of PhCl followed by irradiation generates product 22 in 31% yield (eq. 15). The overall yield is significantly reduced in this case owing to the presence of products arising from some tin-aryl bond fragmentation.
When 22 is further treated with Na metal in liquid ammonia following the same procedures as utilized to form 16 or 21, nucleophile 23 is obtained. Upon photostimulation of 23 in the presence of 4-chlorobiphenyl, product 24, a chiral aromatic organostannyl compound, is obtained in 25% yield (eq. 16).
As mentioned previously, both alkyl-Sn and aryl-Sn bonds can fragment in the presence of Na metal in liq- uid ammonia, decreasing the selectivity of the fragmentation path. Selectivity towards alkyl-Sn bond fragmen- tation at the expense of aryl-Sn bond scission can be improved by exchanging the methyl group for another alkyl group with lower Sn-C bond dissociation energy. In fact, the cleavage of either n-butyltriphenyl tin (BDE (Sn-Bu) = 209 kJ mol
-1) [
20] or benzyltriphenyltin (BDE (Sn-benzyl) = 163 kJ mol
-1) [
22] leads exclusively to Sn-alkyl bond fragmentation.
5. Synthetic Applications
Aryltrialkyl stannanes are valuable intermediates in organic synthesis, and the fact that they can be easily synthesized through the S
RN1 mechanism, opens up important synthetic routes to different reaction schemes. Halodemetallation of aryltrialkyl stannanes has been one such example [
23]. The reaction of Me
3Sn
- ions with aryl chlorides with ulterior addition of iodine in CHCl
3 has been used to obtain aryl iodides from aryl chlorides in very good yields [
17].
For over a decade, the palladium-catalyzed cross-coupling of organotin compounds with carbon electrophiles, known as the Stille reaction [
24], has been a very important tool in organic synthesis [
25]. The reaction of
p-cyanophenyltrimethylstannane
25 [
26] with PhI and Pd(PPh
3)
2Cl
2 as catalyst in DMF (80
oC, 3 h), affords the coupling product 4-biphenycarbonitrile
26 in 81% yield (eq. 17) [
27].
The synthesis of the stannane and the Stille reaction were carried out in a one-pot procedure consisting of irradiating a liquid ammonia solution of
p-chlorobenzonitrile and NaSnMe
3, followed by quenching of the reaction by MeI [
28]. The ammonia was allowed to evaporate, and the residue was taken up in DMF when PhI, and Pd(PPh
3)
2Cl
2 were added. Product
26 was obtained in 63% yield.
As shown before, high yields of disubstitution products can be obtained by the SRN1 mechanism from dichloro arenes and heteroarenes. For instance, when distannane 27a is heated at 80oC in the presence of PhBr and Pd(PPh3)2Cl2 in DMF, m-terphenyl 28a is obtained in 97% isolated yield (eq. 18). Under the same reaction conditions, the distannane 27b affords p-terphenyl 28b in 90% isolated yield.
![Molecules 05 01068 i014]()
The one-pot photoreaction in liquid ammonia of m-dichlorobenzene and NaSnMe3 followed by quenching with MeI, evaporation of the ammonia, and redissolving in DMF in the presence of PhBr and Pd(PPh3)2Cl2 as described above, leads to product 28a in 76% yield. Following the same procedure but using p- dichlorobenzene, product 28b is obtained in 71% yield. Reaction of 2,6-di(trimethylstannyl)pyridine with PhI and Pd(PPh3)2Cl2 affords diphenylated pyridine (72%), along with 2-phenylpyridine (25%). In the one-pot fashion, substrate 2,6-dichloropyridine rendered the diphenylated product in 60% overall yield.
A triphenylation reaction was conducted utilizing the tristannane 9. Upon reaction of 9 with PhI in the presence of catalytic Pd(PPh3)2Cl2 , triphenylated product 29 was obtained in 89% yield (eq. 19). In the one- pot procedure, 1,3,5-trichlorobenzene affords 29 in 61% isolated yield.
The fact that chloroarenes 30 react with Me3Sn- ions under photostimulation to form aryltrimethyl stannanes 31, and that in the Pd-catalyzed Stille reaction with stannanes the reactivity of iodoarenes is much greater than that of chloroarenes, a substrate bearing both leaving groups, Cl and I, will react by the C-I bond (product 32, eq. 20) and not by the C-Cl bond, in a Stille-type reaction. This will allow the remainder leaving group, Cl, to react later in another SRN1-type reaction to form an organostannyl intermediate (product 33) which can ultimately furnish product 34 (eq. 21) by a palladium catalyzed cross-coupling.
![Molecules 05 01068 i016]()
The above sequence, S
RN1 reaction-Pd catalyzed reaction-S
RN1 reaction-Pd catalyzed reaction was investigated in our laboratory in order to foster a methodology to build large molecules. The photo
-stimulated reaction of
p-chlorobenzonitrile
35 with Me
3Sn
- ion affords the stannane
25, which by a Pd (0)-catalyzed reaction with
p-chloroiodobenzene furnishes product
36 in 94% yield (eq. 22). [
15]
By a photostimulated reaction of Me3Sn- ion in the presence of 36, stannane 37 is obtained in 94% yield. A second Pd(0)-catalyzed reaction with 1-iodonaphthalene renders product 38 in 93% yield (eq. 23).
All these results indicate that the SRN1 mechanism is an excellent method to obtain stannanes by the photostimulated reactions of mono-, di- and trichloro arenes with Me3Sn- in liquid ammonia. The stannanes thus obtained can be arylated by further reaction with bromo or iodoarenes through palladium-catalyzed reactions. If the Pd(0)-catalyzed reaction is performed with a chloroiodoarene substrate, the product obtained can be further arylated by a consecutive SRN1-Stille reaction.
6. Conclusions and Summary Remarks
The SRN1 reactions of trimethylstannyl and triphenylstannyl anions with haloarenes are quite versatile. The reaction products are of prominent synthetic relevance and can be utilized as intermediates in important reactions, such as the Stille reaction. Thus, the SRN1 mechanism affords triorganyl stannyl aromatic compounds which otherwise would be synthesized by routes which employ harsher reaction conditions. The sequence SRN1-Pd-catalysis is a powerful synthetic tool, and the scope of the reaction is unlimited owing to the nature of the sequence, i.e. ArX→ArSnR3→ArAr, which can be iteratively repeated when appropriately substituted substrates are chosen.





















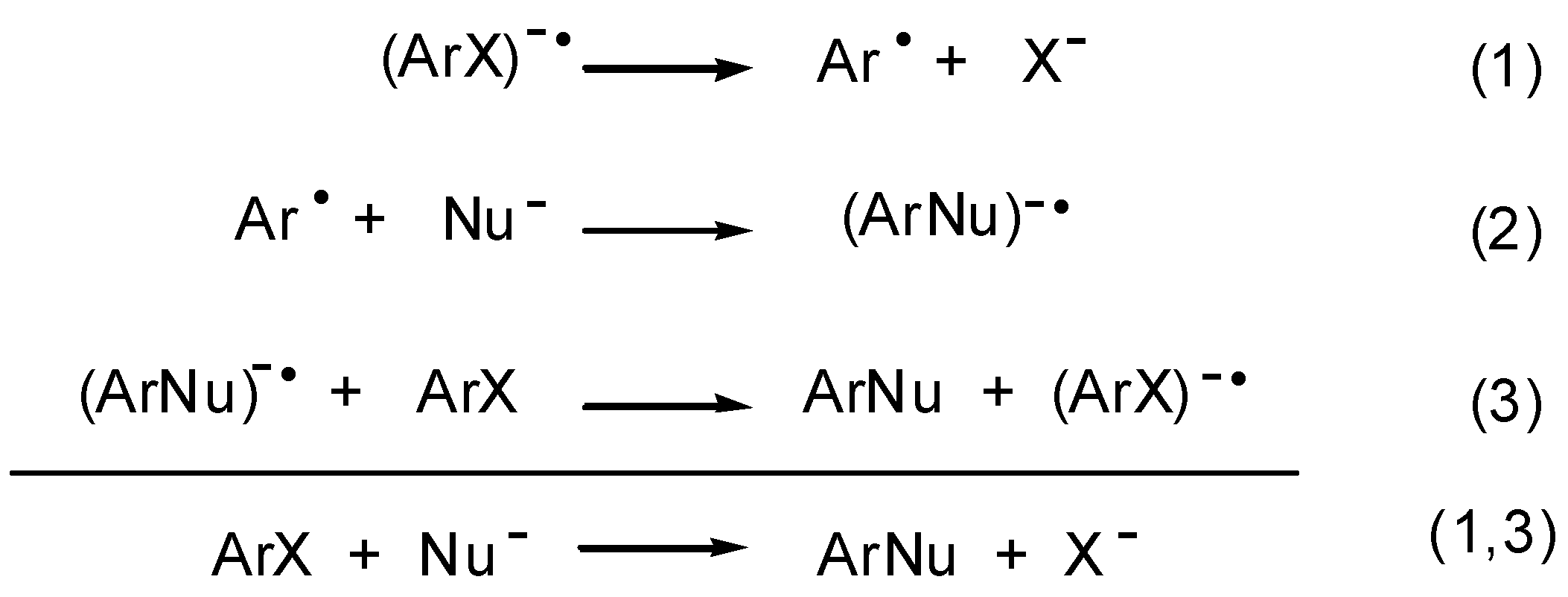{kind=link}

