Conformational Analysis of Thioether Musks Using Density Functional Theory


Abstract
:
1. Introduction
2. Results and Discussion
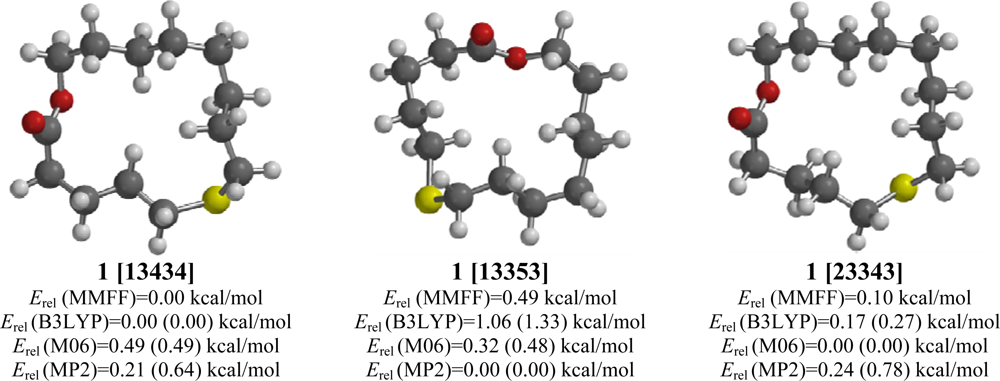
2.1. 6-Thia-14-tetradecanolide (1)
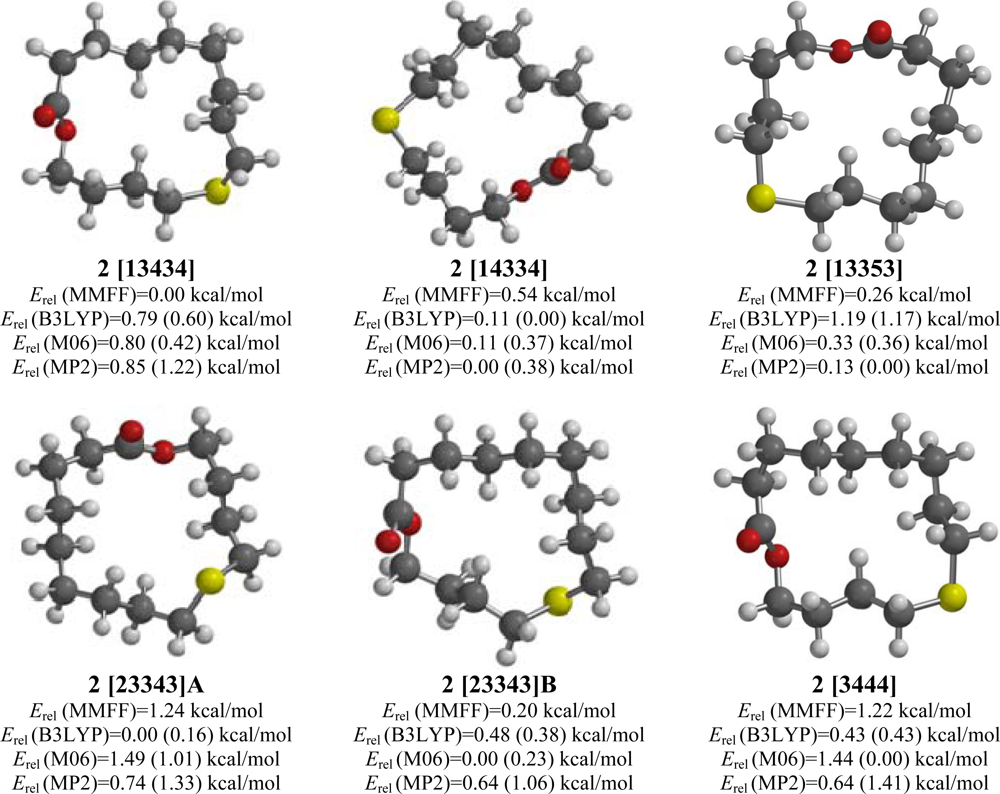
2.2. 10-Thia-14-tetradecanolide (2)
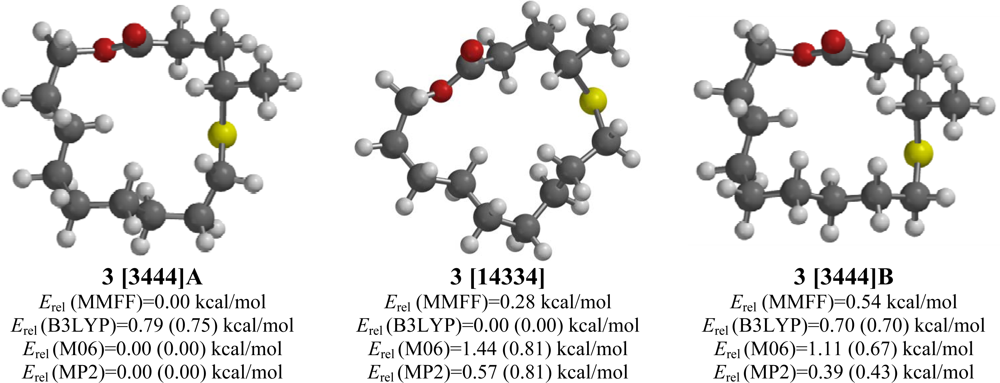
2.3. 4-Methyl-5-thia-14-tetradecananolide (3)
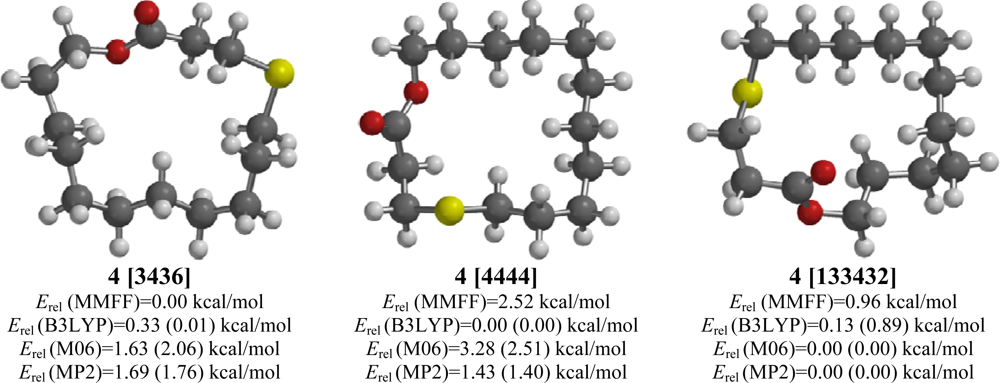
2.4. 4-Thia-15-pentadecanolide (4)
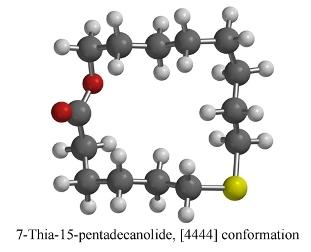
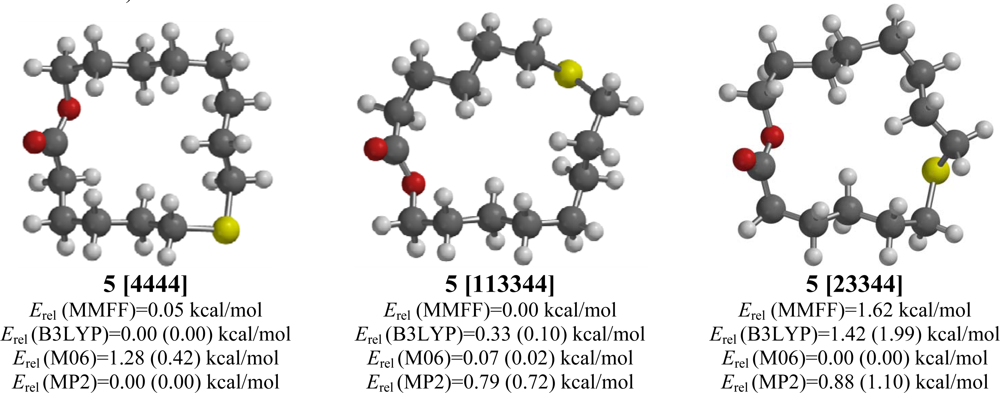
2.5. 7-Thia-15-pentadecanolide (5)
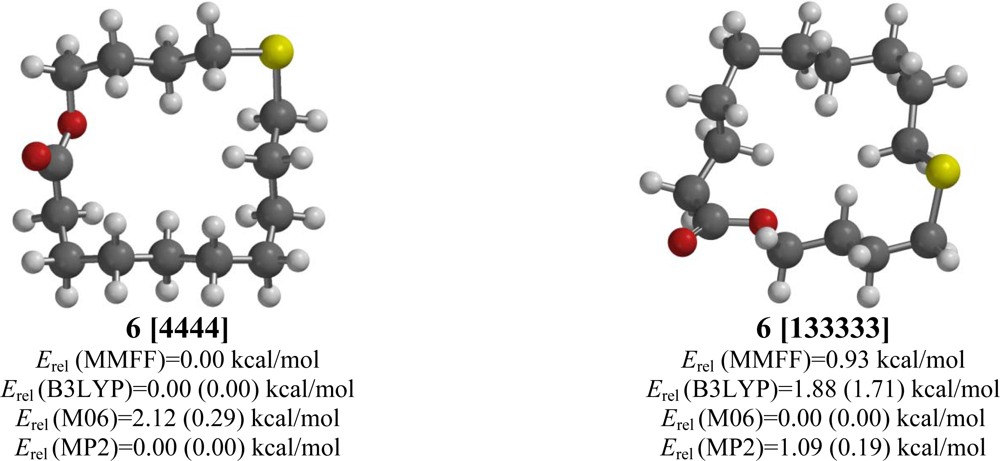
2.6. 11-Thia-15-pentadecanolide (6)
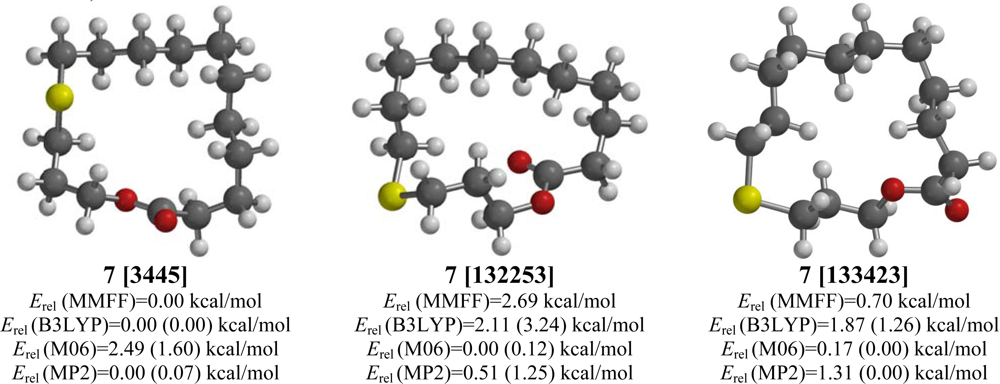
2.7. 12-Thia-15-pentadecanolide (7)
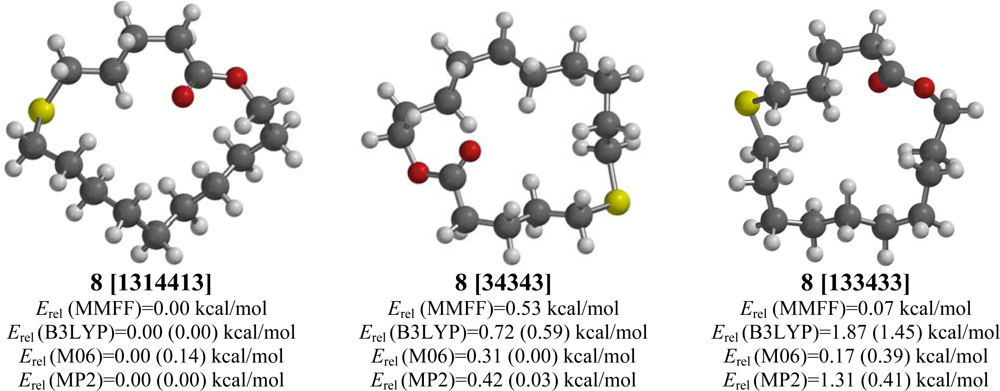
2.8. 6-Thia-16-hexadecanolide (8)
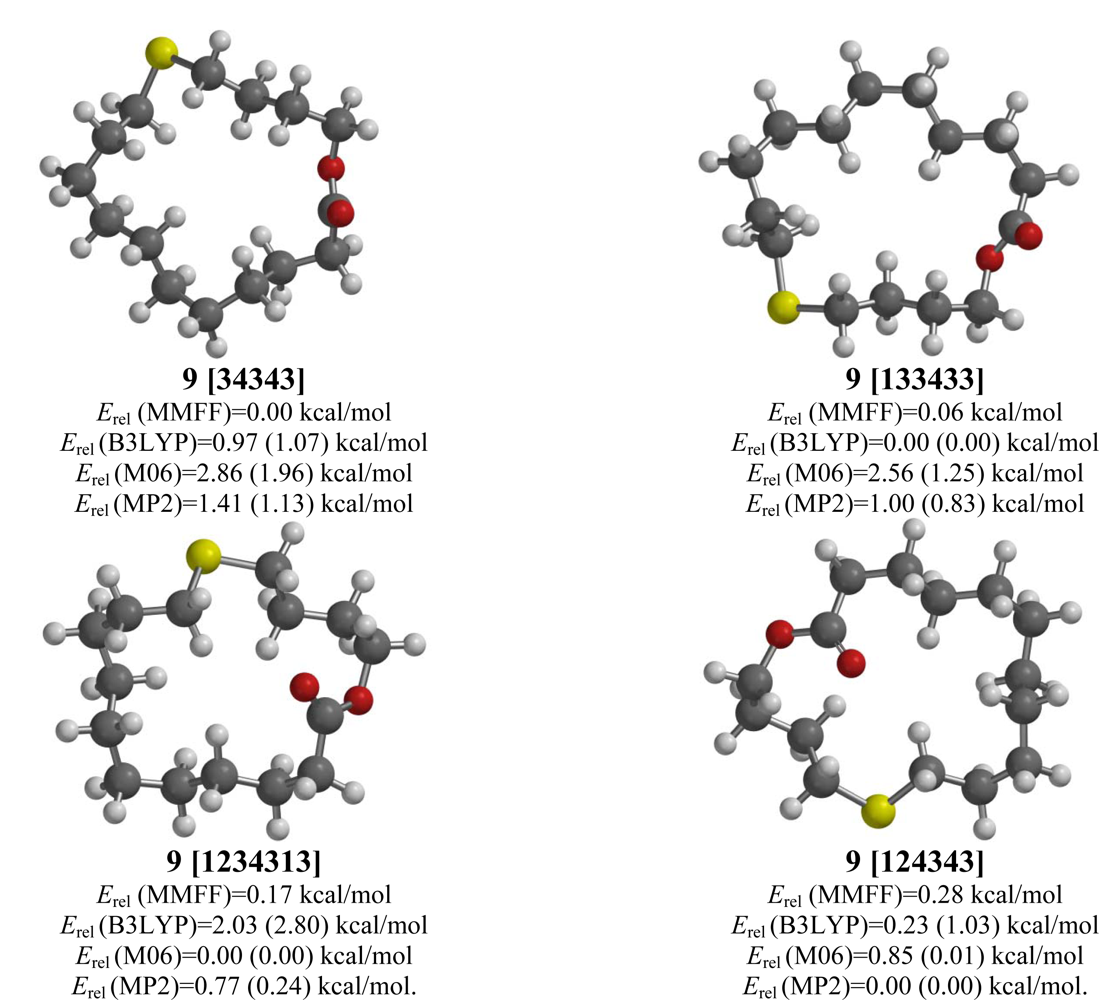
2.9. 12-Thia-16-hexadecanolide (9)
3. Computational Methods
4. Conclusions
References and Notes
- Dos Santos, HF; Rocha, WR; de Almeida, WB. On the evaluation of thermal corrections to gas phase ab initio relative energies: Implications to the conformational analysis study of cyclooctane. Chem. Phys 2002, 280, 31–42. [Google Scholar]
- Suvire, FD; Santagata, LN; Bombasaro, JA; Enriz, RD. Dynamics of flexible cycloalkanes. Ab initio and DFT study of the conformational energy hypersurface of cyclononane. J. Comput. Chem 2006, 27, 188–202. [Google Scholar]
- Setzer, WN. A theoretical investigation into the formation of sesquiterpene episulfides. J. Sulfur Chem 2009, 30, 119–127. [Google Scholar]
- Schreiner, PR; Fokin, AA; Pascal, RA; de Meijere, A. Many density functional theory approaches fail to give reliable large hydrocarbon isomer energy differences. Org. Lett 2006, 8, 3635–3638. [Google Scholar]
- Wodrich, MD; Corminboeuf, C; Schreiner, PR; Fokin, AA; Schleyer, PvR. How accurate are DFT treatments of organic energies? Org. Lett 2007, 9, 1851–1854. [Google Scholar]
- Theimer, ET; Davies, JT. Olfaction, musk odor, and molecular properties. J. Agric. Food Chem 1967, 15, 6–14. [Google Scholar]
- Lopes, D; Strobl, H; Kolodziejczyk, P. 14-Methylpentadecano-15-lactone (muscolide): A new macrocyclic lactone from the oil of Angelica archangelica L. Chem. Biodivers 2004, 1, 1880–1887. [Google Scholar]
- Allen, CFH; VanAllan, JA. Some macrocyclic oxalactones and related substances. J. Org. Chem 1949, 14, 754–760. [Google Scholar]
- Goeke, A. Sulfur-containing odorants in fragrance chemistry. Sulfur Rep 2002, 23, 243–278. [Google Scholar]
- Halgren, TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF 94. J. Comput. Chem 1996, 17, 490–519. [Google Scholar]
- Becke, AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Lee, C; Yang, W; Parr, RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Zhao, Y; Truhlar, DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accts 2008, 120, 215–241. [Google Scholar]
- Dale, J. Exploratory calculations of medium and large rings. Acta Chem. Scand 1973, 27, 1115–1129. [Google Scholar]
- Noe, EA; Pawar, DM; Fronczek, FR. Cyclopentadecanone. Acta Crystallogr 2008, C64, o67–o68. [Google Scholar]
- Wolf, RE; Hartman, JR; Storey, JME; Foxman, BM; Cooper, SR. Crown thioether chemistry: Structural and conformational studies of tetrathia-12-crown-4, pentathia-15-crown-5, and hexathia-18-crown-6. Implications for ligand design. J. Am. Chem. Soc 1987, 109, 4328–4335. [Google Scholar]
- Desper, JM; Powell, DR; Gellman, SH. Molecular structure and conformation of two acyclic polythioethers: Implications for the design of heavy metal chelators. J. Am. Chem. Soc 1990, 112, 4321–4324. [Google Scholar]
- Huisgen, R; Ott, H. Die configuration der carbonestergruppe und die sondereigenshaften der lactone. Tetrahedron 1959, 6, 253–267. [Google Scholar]
- Dale, J. Macrocyclic compounds. Part III. Conformations of cycloalkanes and other flexible macrocycles. J Chem Soc 1963, 93–111. [Google Scholar]
- Pinkus, AG; Lin, EY. Electric dipole moment studies of carboxylic ester conformations: Alkyl acetates, benzoates, 2,4,6-trimethylbenzoates and 2,3,5,6-tetramethylbenzoates. J. Mol. Struct 1975, 24, 9–26. [Google Scholar]
- Setzer, WN; Cacioppo, EL; Grant, GJ; Glass, RS. The crystal and molecular structure of 1,4,7,11,14,17-hexathiacycloeicosane-9,19-dione: A macrocyclic diketo-hexathioether. Phosphorus Sulfur Silicon Relat. Elem 1989, 45, 223–229. [Google Scholar]
- Setzer, WN; Grant, GJ; Meehan, EM; Lee, SD; Kirk, RM; Yang, CR; Wu, CK. The crystal structure of the macrocyclic hexathioether 1,4,7,11,14,17-hexathiacycloeicosane: Implications for metal complexation. J. Incl. Phenom. Macrocycl. Chem 1999, 35, 649–656. [Google Scholar]
- Anet, FAL; Cheng, AK. Conformation of cyclohexadecane. J. Am. Chem. Soc 1975, 97, 2420–2424. [Google Scholar]
- Zhang, R; Mattice, WL. Molecular dynamics study of the conformational properties of cyclohexadecane. J. Chem. Phys 1993, 98, 9888–9894. [Google Scholar]
- Sanders, M; Houk, KN; Wu, YD; Still, WC; Lipton, M; Chang, G; Guida, WC. Conformations of cycloheptadecane. A comparison of methods for conformational searching. J. Am. Chem. Soc 1993, 112, 1419–1427. [Google Scholar]
- Csonka, GI. Proper basis set for quantum mechanical studies of potential energy surfaces of carbohydrates. Theochem 2002, 584, 1–4. [Google Scholar]
- Csonka, GI; French, AD; Johnson, GP; Stortz, CA. Evaluation of density functionals and basis sets for carbohydrates. J. Chem. Theor. Comput 2009, 5, 679–692. [Google Scholar]
- Lynch, BJ; Zhao, Y; Truhlar, DG. Effectiveness of diffuse basis functions for calculating relative energies by density functional theory. J. Phys. Chem. A 2003, 107, 1384–1388. [Google Scholar]
- Setzer, WN; Liou, SY; Easterling, GE; Simmons, RC; Gullion, LM; Meehan, EJ; Grant, GJ; Gray, GM. Synthesis and structural studies of hydrophilic mesocyclic trithioethers. Heteroatom Chem 1998, 9, 123–128. [Google Scholar]
- SPARTAN ’08 for Windows; Wavefunction, Inc: Irvine, CA, USA, 2008.
- Hehre, WJ; Radom, L; Schleyer, PvR. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Chambers, CC; Hawkins, GD; Cramer, CJ; Truhlar, DG. Model for aqueous solvation based on class IV atomic charges and first solvation shell effects. J. Phys. Chem 1996, 100, 16385–16398. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths (Å) | X-ray | 10 | M06 | 1 [13434] | 5 [4444] | ||
|---|---|---|---|---|---|---|---|
| B3LYP | B3LYP | M06 | B3LYP | M06 | |||
| CH2–CH2 | 1.522 | 1.539 | 1.525 | 1.536 | 1.522 | 1.536 | 1.522 |
| CH2–S (endodentate) | 1.816 | 1.845 | 1.833 | 1.843 | 1.829 | --- | --- |
| CH2–S (exodentate) | 1.798 | 1.839 | 1.827 | --- | --- | 1.841 | 1.828 |
| C=O | 1.206 | 1.211 | 1.206 | 1.214 | 1.209 | 1.213 | 1.209 |
| C–O | 1.316 | 1.356 | 1.346 | 1.353 | 1.344 | 1.354 | 1.345 |
| Bond Angles (deg) | |||||||
| CH2–CH2–CH2 | 112.5 | 112.9 | 112.4 | 114.12 | 113.48 | 113.95 | 113.32 |
| CH2–CH2–S | 113.9 | 114.4 | 113.8 | 116.0 | 115.8 | 115.8 | 115.4 |
| CH2–S–CH2 (endodentate) | 100.1 | 100.1 | 98.4 | 103.1 | 101.6 | --- | --- |
| CH2–S–CH2 (exodentate) | 101.1 | 101.4 | 100.5 | --- | --- | 102.9 | 101.3 |
| O–C=O | 122.7 | 122.2 | 122.4 | 124.0 | 124.3 | 123.9 | 123.9 |
| Compound (Conformation) | Dipole (D) | Erel (kcal/mol)a | |||
|---|---|---|---|---|---|
| MMFF | B3LYP/6–31+G* | M06/6–31+G* | MP2/6–311+G** | ||
| 6-Thia-14-tetradecanolide (1) | |||||
| [13434] | 0.88 | 0.00 | 0.00 (0.00) | 0.49 (0.00) | 0.64 (0.58) |
| [13353] | 1.90 | 0.49 | 1.33 (1.40) | 0.48 (0.04) | 0.00 (0.00) |
| [23343] | 1.15 | 0.10 | 0.27 (0.74) | 0.00 (0.00) | 0.78 (1.19) |
| 10-Thia-14-tetradecanolide (2) | |||||
| [13434] | 0.76 | 0.00 | 0.60 (0.73) | 0.42 (0.70) | 1.22 (1.38) |
| [13353] | 1.94 | 0.26 | 1.17 (1.14) | 0.36 (0.51) | 0.00 (0.00) |
| [23343]A | 3.11 | 1.24 | 0.16 (0.54) | 1.01 (1.68) | 1.33 (1.74) |
| [23343]B | 0.77 | 0.20 | 0.38 (0.95) | 0.23 (1.01) | 1.06 (1.65) |
| [14334] | 1.73 | 0.54 | 0.00 (0.00) | 0.37 (0.56) | 0.38 (0.41) |
| [3444] | 1.35 | 1.22 | 0.43 (0.20) | 0.00 (0.00) | 1.41 (1.21) |
| 4-Methyl-5-thia-14-tetradecananolide (3) | |||||
| [3444]A | 0.91 | 0.00 | 0.75 (0.89) | 0.00 (0.00) | 0.00 (0.00) |
| [3444]B | 0.97 | 0.54 | 0.70 (0.65) | 0.67 (0.41) | 0.43 (0.24) |
| [14334] | 1.60 | 0.28 | 0.00 (0.00) | 0.81 (0.83) | 0.81 (0.67) |
| 4-Thia-15-pentadecanolide (4) | |||||
| [4444] | 3.19 | 2.52 | 0.00 (0.00) | 2.51 (2.22) | 1.40 (0.94) |
| [3436] | 3.51 | 0.00 | 0.01 (0.01) | 2.06 (1.90) | 1.76 (1.30) |
| [133432] | 0.65 | 0.96 | 0.89 (1.34) | 0.00 (0.00) | 0.00 (0.00) |
| 7-Thia-15-pentadecanolide (5) | |||||
| [4444] | 2.43 | 0.05 | 0.00 (0.00) | 0.42 (0.00) | 0.00 (0.00) |
| [23344] | 1.55 | 1.62 | 1.99 (3.01) | 0.00 (0.77) | 1.10 (2.12) |
| [113344] | 1.54 | 0.00 | 0.10 (0.75) | 0.02 (0.21) | 0.72 (1.36) |
| 11-Thia-15-pentadecanolide (6) | |||||
| [4444] | 1.81 | 0.00 | 0.00 (0.00) | 0.29 (0.00) | 0.00 (0.00) |
| [133333] | 2.25 | 0.93 | 1.71 (2.82) | 0.00 (0.81) | 0.19 (1.31) |
| 12-Thia-15-pentadecanolide (7) | |||||
| [3445] | 3.27 | 0.00 | 0.00 (0.00) | 1.60 (2.22) | 0.07 (0.57) |
| [132253] | 2.49 | 2.69 | 3.24 (3.16) | 0.12 (0.50) | 1.25 (1.67) |
| [133423] | 2.44 | 0.70 | 1.26 (0.76) | 0.00 (0.00) | 0.00 (0.00) |
| 6-Thia-16-hexadecanolide (8) | |||||
| [34343] | 2.87 | 0.53 | 0.59 (0.46) | 0.00 (0.00) | 0.03 (0.00) |
| [133433] | 3.20 | 0.07 | 1.45 (1.21) | 0.39 (0.49) | 0.41 (0.26) |
| [1314413] | 2.24 | 0.00 | 0.00 (0.00) | 0.14 (0.38) | 0.00 (0.11) |
| 12-Thia-16-hexadecanolide (9) | |||||
| [34343] | 1.77 | 0.00 | 1.07 (0.93) | 1.96 (1.42) | 1.13 (0.61) |
| [133433] | 1.51 | 0.06 | 0.00 (0.00) | 1.25 (0.56) | 0.83 (0.16) |
| [124343] | 2.76 | 0.28 | 1.03 (1.71) | 0.01 (0.00) | 0.00 (0.00) |
| [1234313] | 2.78 | 0.17 | 2.80 (3.72) | 0.00 (0.22) | 0.24 (0.48) |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Setzer, W.N. Conformational Analysis of Thioether Musks Using Density Functional Theory. Int. J. Mol. Sci. 2009, 10, 3488-3501. https://doi.org/10.3390/ijms10083488
Setzer WN. Conformational Analysis of Thioether Musks Using Density Functional Theory. International Journal of Molecular Sciences. 2009; 10(8):3488-3501. https://doi.org/10.3390/ijms10083488
Chicago/Turabian StyleSetzer, William N. 2009. "Conformational Analysis of Thioether Musks Using Density Functional Theory" International Journal of Molecular Sciences 10, no. 8: 3488-3501. https://doi.org/10.3390/ijms10083488
APA StyleSetzer, W. N. (2009). Conformational Analysis of Thioether Musks Using Density Functional Theory. International Journal of Molecular Sciences, 10(8), 3488-3501. https://doi.org/10.3390/ijms10083488