Molecular Modeling Studies of 4,5-Dihydro-1H-pyrazolo[4,3-h] quinazoline Derivatives as Potent CDK2/Cyclin A Inhibitors Using 3D-QSAR and Docking

Abstract
:1. Introduction
2. Results and Discussion
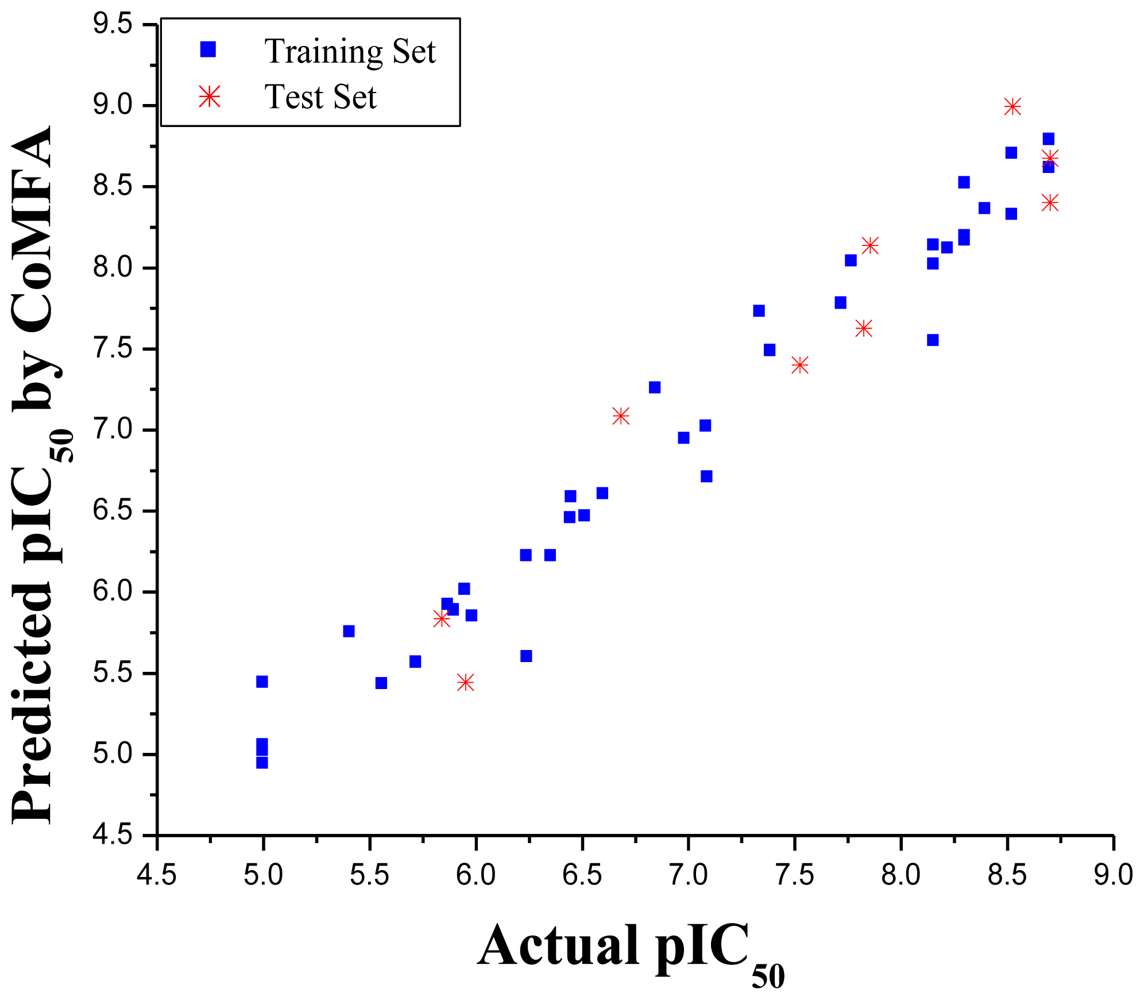
2.1. CoMFA Model
2.2. CoMSIA Model
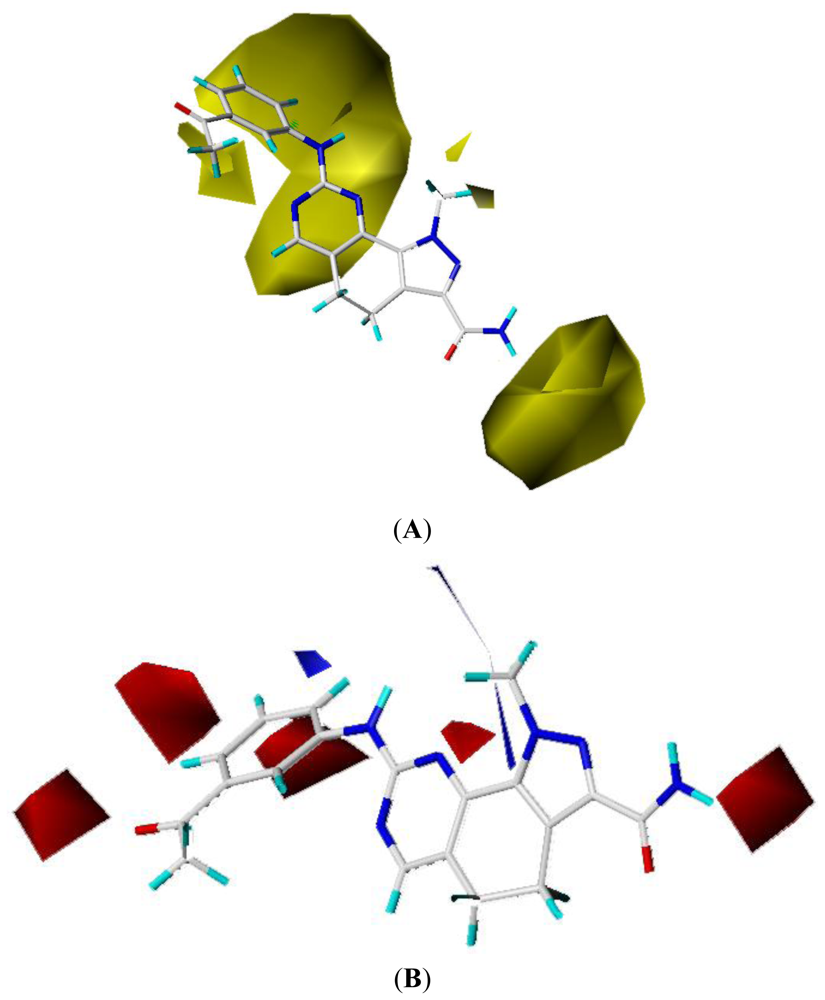
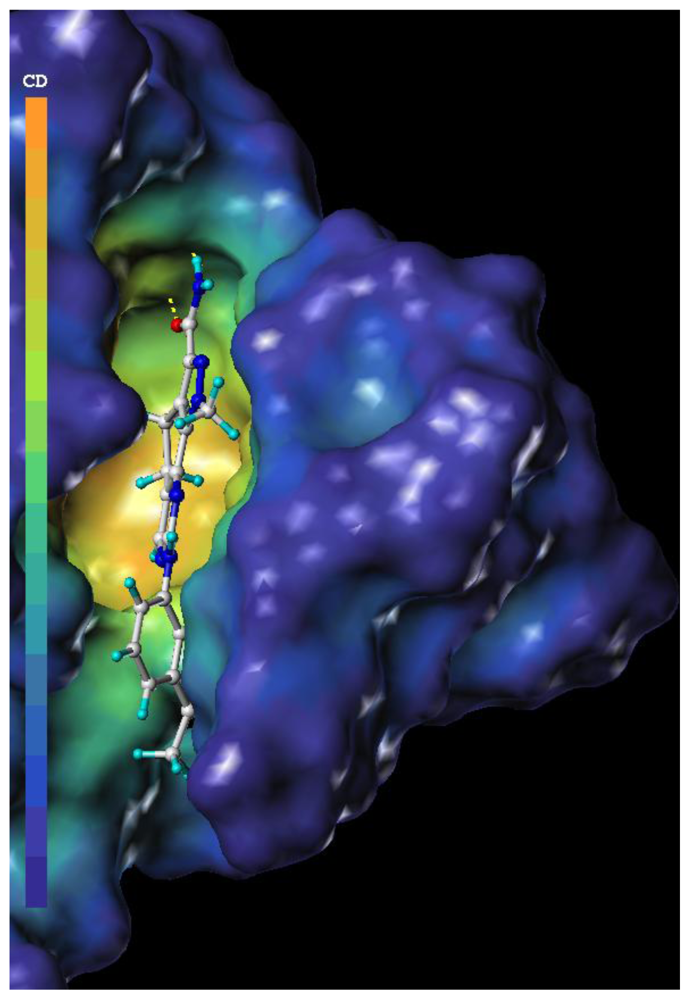
2.3. CoMFA Contour Maps
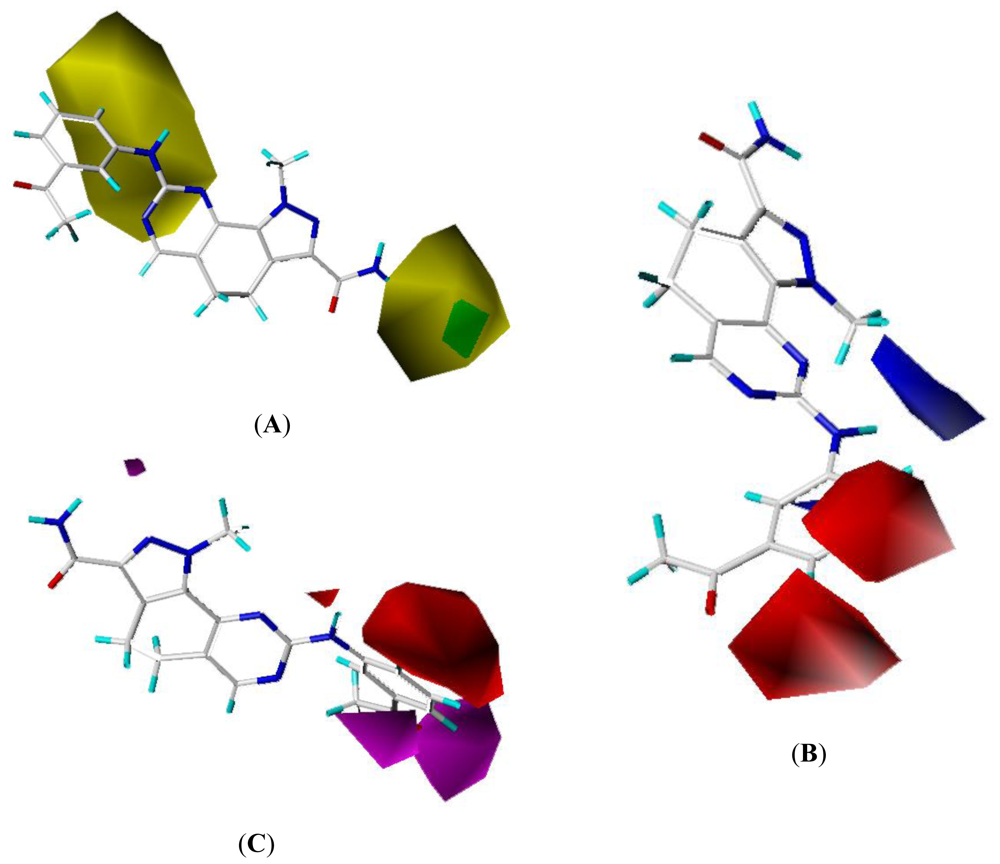
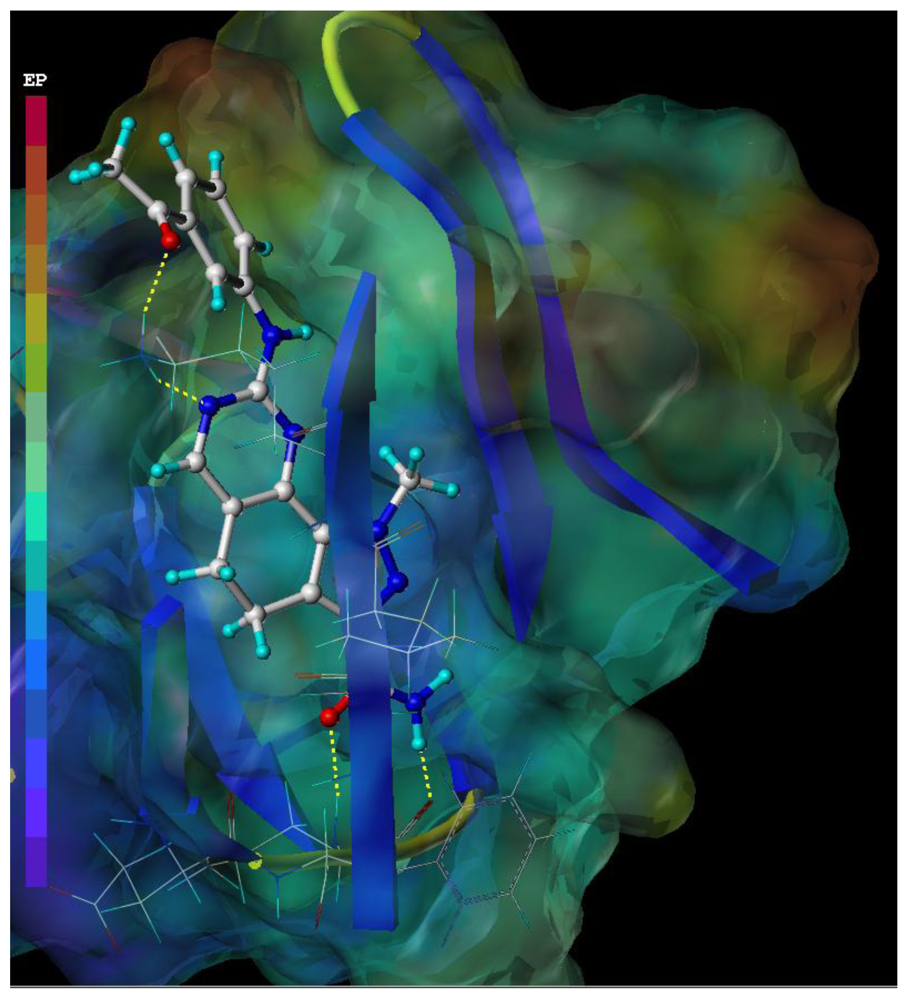
2.4. CoMSIA Contour Maps
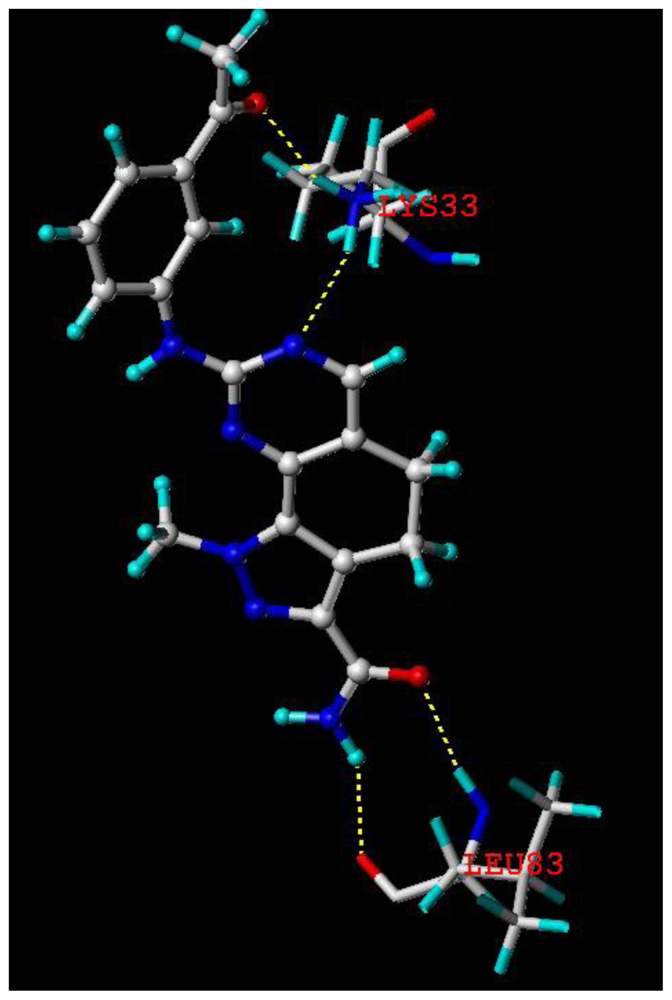
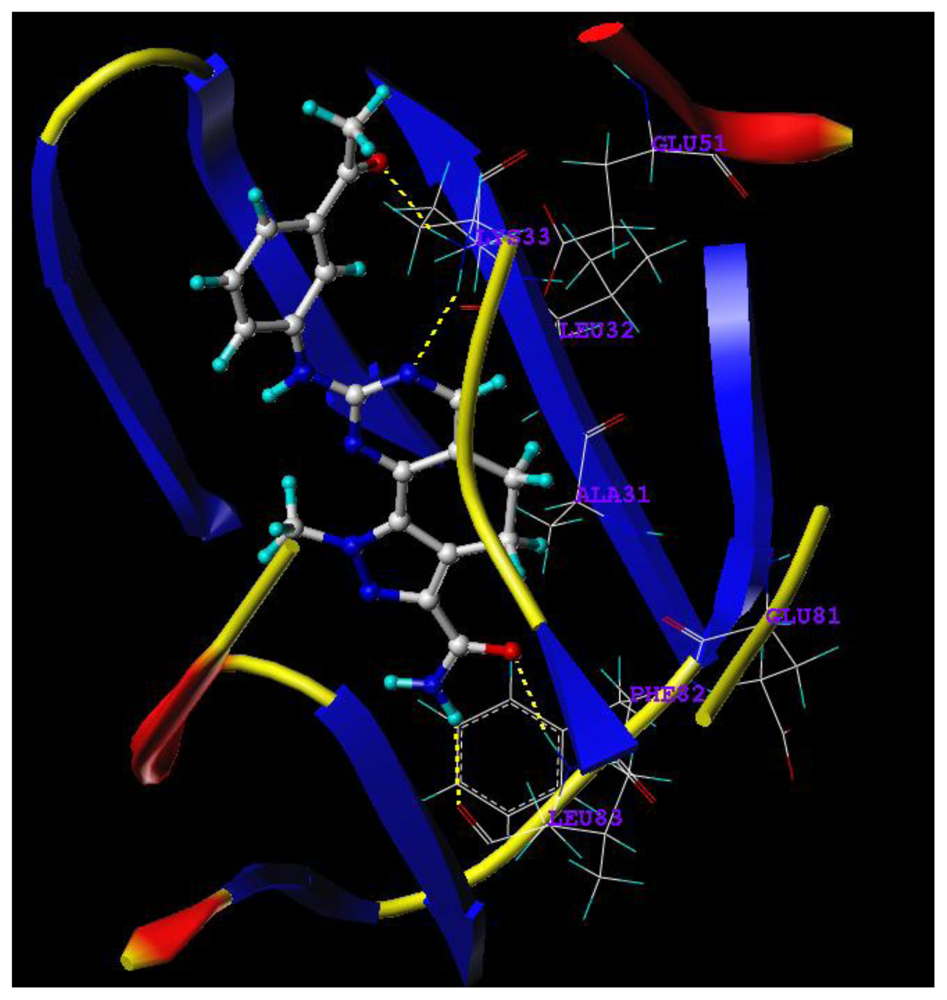
2.5. Docking Analysis
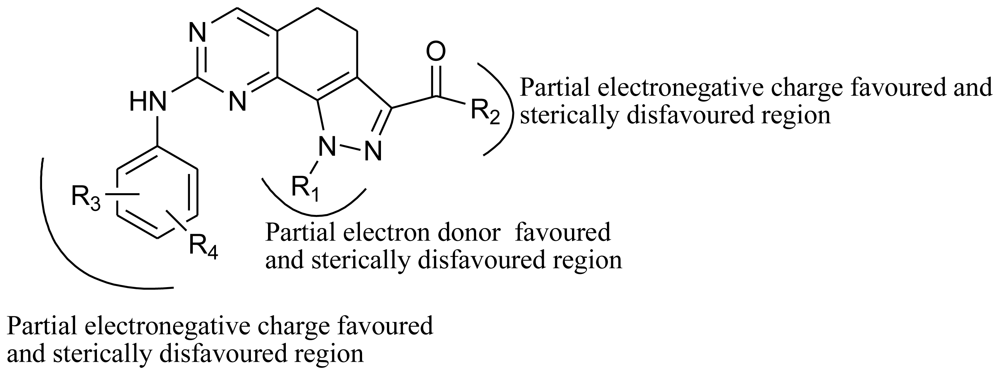
2.6. Design of New Molecules Based on COMFA, CoMSIA and Docking Studies
3. Materials and Methods
3.1. Data Sets
3.2. Molecular Modeling and Alignment
3.3. CoMFA and CoMSIA Modeling
3.4. Partial Least Squares (PLS) Analysis
3.5. Predictive Correlation Co-Efficient (r2pred)
3.6. Molecular Docking
4. Conclusion
Acknowledgement
References
- Renfrey, S; Featherstone, J. From the analyst’s couch: Structural proteomics. Nat. Rev. Drug Discov 2002, 1, 175–176. [Google Scholar]
- Cohen, P. Protein kinases: The major drug targets of the twenty-first century? Nat. Rev. Drug Discov 2002, 1, 309–315. [Google Scholar]
- Noble, MEM; Endicott, JA; Johnson, LN. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science 2004, 303, 1800–1805. [Google Scholar]
- Cruz, JC; Tsai, LH. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol. Med 2004, 10, 452–458. [Google Scholar]
- Smith, PD; O’Hare, MJ; Park, DS. CDKs: Taking on a role as mediators of dopaminergic loss in Parkinson’s disease. Trends Mol. Med 2004, 10, 445–451. [Google Scholar]
- Zhang, M; Li, J; Chakrabarty, P; Bu, B; Vincent, I. Cyclin-dependent kinase inhibitors attenuate protein hyperphosphorylation, cytoskeletal lesion formation, and motor defects in Niemann-Pick type C mice. Am. J. Pathol 2004, 165, 843–852. [Google Scholar]
- Wang, J; Liu, S; Fu, Y; Wang, JH; Lu, Y. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat. Neurosci 2003, 6, 1039–1047. [Google Scholar]
- Di Giovanni, S; Movsesyan, V; Ahmed, F; Cernak, I; Schinelli, S; Stoica, B; Faden, AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. USA 2005, 102, 8333–8335. [Google Scholar]
- Dai, Y; Grant, S. Cyclin-dependent kinase inhibitors. Cur. Opin. Pharmacol 2003, 3, 363–370. [Google Scholar]
- Ficher, PM; Gianella-Borradori, A. CDK inhibitors in clinical development for the treatment of cancer. Exp. Opin. Invest. Drugs 2003, 12, 955–970. [Google Scholar]
- Durdagi, S; Papadopoulos, MG; Papahatjis, DP; Mavromoustakos, T. Combined 3D QSAR and molecular docking studies to reveal novel cannabinoid ligands with optimum binding activity. Bioorg. Med. Chem. Lett 2007, 17, 6754–6763. [Google Scholar]
- Durdagi, S; Mavromoustakos, T; Papadopoulos, MG. 3D QSAR CoMFA/CoMSIA, molecular docking and molecular dynamics studies of fullerene-based HIV-1 PR inhibitors. Bioorg. Med. Chem. Lett 2008, 18, 6283–6289. [Google Scholar]
- Mascarenhas, NM; Ghoshal, N. An efficient tool for identifying inhibitors based on 3D-QSAR and docking using feature-shape pharmacophore of biologically active conformation—A case study with CDK2/CyclinA. Eur. J. Med. Chem 2008, 43, 2807–2818. [Google Scholar]
- Singh, SK; Dessalew, N; Bharatam, PV. 3D-QSAR CoMFA study on indenopyrazole derivatives as cyclin dependent kinase 4 (CDK4) and cyclin dependent kinase 2 (CDK2) inhibitors. Eur. J. Med. Chem 2006, 41, 1310–1319. [Google Scholar]
- Beria, I; Ballinari, D; Bertrand, JA; Borghi, D; Bossi, RT; Brasca, MG; Cappella, P; Caruso, M; Ceccarelli, W; Ciavolella, A; Cristiani, C; Croci, V; Ponti, AD; Fachin, G; Ferguson, RD; Lansen, J; Moll, JK; Pesenti, E; Posteri, H; Perego, R; Rocchetti, M; Storici, P; Volpi, D; Valsasina, B. Identification of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline Derivatives as a New Class of Orally and Selective Polo-Like Kinase 1 Inhibitors. J. Med. Chem 2010, 53, 3532–3551. [Google Scholar]
- Sybyl 8.1. Tripos Inc: St. Louis, MO, USA, 2008. Available online: http://www.tripos.com.
- Yang, ZQ; Sun, PH. 3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach. Int. J. Mol. Sci 2007, 8, 714–722. [Google Scholar]
- Song, QL; Sun, PH; Chen, WM. Exploring 3D-QSAR for Ketolide Derivatives as Antibacterial Agents Using CoMFA and CoMSIA. Lett. Drug Des. Discov 2010, 7, 149–159. [Google Scholar]
- Politi, A; Durdagi, S; Moutevelis-Minakakis, P; Kokotos, G; Papadopoulos, MG; Mavromoustakos, T. Application of 3D QSAR CoMFA/CoMSIA and in silico docking studies on novel renin inhibitors against cardiovascular diseases. Eur. J. Med. Chem 2009, 44, 3703–3711. [Google Scholar]
- Cichero, E; Cesarini, S; Spallarossa, A; Mosti, L; Fossa, P. Acylthiocarbamates as non-nucleoside HIV-1 reverse transcriptase inhibitors: dockinig studies and ligand-based CoMFA and CoMSIA analyses. J. Mol. Model 2009, 15, 871–884. [Google Scholar]
- Cichero, E; Cesarini, S; Fossa, P; Spallarossa, A; Mosti, L. Thiocarbamates as non-nucleoside HIV-1 reverse transcriptase inhibitors: Docking-based CoMFA and CoMSIA analyses. Eur. J. Med. Chem 2009, 44, 2059–2070. [Google Scholar]
- Pan, X; Tan, N; Zeng, G; Huang, H; Yan, H. 3D QSAR studies on ketoamides of human cathepsin K inhibitors based on two different alignment methods. Eur. J. Med. Chem 2010, 45, 667–681. [Google Scholar]
- Cichero, E; Cesarini, S; Mosti, L; Fossa, P. CoMFA and CoMSIA analyses on 4-oxo-1,4-dihydroquinoline and 4-oxo-1,4-dihydro-1,5-,-1,6- and -1,8-naphthyridine derivatives as selective CB2 receptor agonists. J. Mol. Model 2010, 16, 677–691. [Google Scholar]
- Cichero, E; Cesarini, S; Spallarossa, A; Mosti, L; Fossa, P. Computational studies of the binding mode and 3D-QSAR analyses of symmetric formimidoester disulfides: A new class of non-nucleoside HIV-1 reverse transcriptase inhibitor. J. Mol. Model 2009, 15, 357–367. [Google Scholar]
- Aher, YD; Agrawal, A; Bharatam, PV; Garg, P. 3D-QSAR studies of substituted 1-(3,3-diphenylpropyl)-piperidinyl amides and ureas as CCR5 receptor antagonists. J. Mol. Model 2007, 13, 519–529. [Google Scholar]
- Sun, J; Cai, S; Yan, N; Mei, H. Docking and 3D-QSAR studies of influenza neuraminidase inhibitors using three-dimensional holographic vector of atomic interaction field analysis. Eur. J. Med. Chem 2010, 45, 1008–1014. [Google Scholar]
- Jain, AN. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem 2003, 46, 499–511. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Statistics | CoMFA | CoMSIA |
|---|---|---|
| r2cva | 0.747 | 0.518 |
| r2 b | 0.970 | 0.934 |
| ONCc | 5 | 6 |
| SEEd | 0.225 | 0.339 |
| F valuee | 206.080 | 72.528 |
| r2predf | 0.942 | 0.931 |
| Field contribution | ||
| Steric | 0.599 | 0.373 |
| Electrostatic | 0.401 | 0.472 |
| Hydrophobic | - | - |
| H-bond Donor | - | - |
| H-bond Acceptor | - | 0.155 |
| Compd. No. | pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Actual | Pred. | Res. | Pred. | Res. | |
| 1* | 8.699 | 8.402 | 0.297 | 8.405 | 0.294 |
| 2 | 8.301 | 8.166 | 0.135 | 8.535 | −0.234 |
| 3* | 7.824 | 7.629 | 0.195 | 7.555 | 0.269 |
| 4 | 8.699 | 8.613 | 0.086 | 8.534 | 0.165 |
| 5 | 8.155 | 7.547 | 0.608 | 7.440 | 0.715 |
| 6 | 7.337 | 7.727 | −0.390 | 7.943 | −0.606 |
| 7 | 6.983 | 6.942 | 0.041 | 7.101 | −0.118 |
| 8* | 7.523 | 7.402 | 0.121 | 7.315 | 0.208 |
| 9 | 6.600 | 6.602 | −0.002 | 6.854 | −0.254 |
| 10 | 8.523 | 8.701 | −0.178 | 8.615 | −0.092 |
| 11 | 7.721 | 7.778 | −0.057 | 7.581 | 0.140 |
| 12 | 7.092 | 6.704 | 0.388 | 6.720 | 0.372 |
| 13 | 5.409 | 5.750 | −0.341 | 5.658 | −0.249 |
| 14 | 6.241 | 6.219 | 0.022 | 6.183 | 0.058 |
| 15 | 6.514 | 6.466 | 0.048 | 6.684 | −0.170 |
| 16 | 8.222 | 8.115 | 0.107 | 8.210 | 0.012 |
| 17* | 8.523 | 8.994 | −0.471 | 8.428 | 0.095 |
| 18 | 5.899 | 5.887 | 0.012 | 5.851 | 0.048 |
| 19 | 8.699 | 8.788 | −0.089 | 8.774 | −0.075 |
| 20* | 8.699 | 8.676 | 0.023 | 8.819 | −0.120 |
| 21 | 7.387 | 7.485 | −0.098 | 7.345 | 0.042 |
| 22 | 8.301 | 8.521 | −0.220 | 8.275 | 0.026 |
| 23 | 8.398 | 8.360 | 0.038 | 8.424 | −0.026 |
| 24* | 6.680 | 7.088 | −0.408 | 6.063 | 0.617 |
| 25 | 8.301 | 8.194 | 0.107 | 8.310 | −0.009 |
| 26* | 7.854 | 8.137 | −0.283 | 8.305 | −0.451 |
| 27 | 8.523 | 8.324 | 0.199 | 8.185 | 0.338 |
| 28 | 6.848 | 7.253 | −0.405 | 7.573 | −0.725 |
| 29 | 7.770 | 8.037 | −0.267 | 7.844 | −0.074 |
| 30 | 8.155 | 8.136 | 0.019 | 7.719 | 0.436 |
| 31 | 6.446 | 6.455 | −0.009 | 6.656 | −0.210 |
| 32* | 5.839 | 5.837 | 0.002 | 5.429 | 0.410 |
| 33 | 5.561 | 5.430 | 0.131 | 5.515 | 0.046 |
| 34 | 5.951 | 6.011 | −0.060 | 5.844 | 0.107 |
| 35 | 5.871 | 5.920 | −0.049 | 5.754 | 0.117 |
| 36 | 5.721 | 5.564 | 0.157 | 5.532 | 0.189 |
| 37 | 6.243 | 5.598 | 0.645 | 6.139 | 0.104 |
| 38 | 6.355 | 6.220 | 0.135 | 5.995 | 0.360 |
| 39 | 5.000 | 5.018 | −0.018 | 4.846 | 0.154 |
| 40 | 5.000 | 5.439 | −0.439 | 5.833 | −0.833 |
| 41 | 8.155 | 8.019 | 0.136 | 7.869 | 0.286 |
| 42 | 5.984 | 5.849 | 0.135 | 5.709 | 0.275 |
| 43* | 5.950 | 5.443 | 0.507 | 5.979 | −0.029 |
| 44 | 5.000 | 4.940 | 0.060 | 5.326 | −0.326 |
| 45 | 5.000 | 5.053 | −0.053 | 5.040 | −0.040 |
| 46 | 7.086 | 7.019 | 0.067 | 6.823 | 0.263 |
| 47 | 6.450 | 6.583 | −0.133 | 6.456 | −0.006 |
| r2cv | r2 | ONC | SEE | F value | r2pred | |
|---|---|---|---|---|---|---|
| S + E | 0.593 | 0.943 | 6 | 0.315 | 85.009 | 0.965 |
| S + E + H | 0.415 | 0.947 | 6 | 0.303 | 92.344 | 0.887 |
| S + E + D | 0.449 | 0.940 | 6 | 0.322 | 80.894 | 0.937 |
| S + E + A* | 0.518 | 0.934 | 6 | 0.339 | 72.528 | 0.931 |
| H + D + A | 0.276 | 0.637 | 2 | 0.746 | 30.677 | 0.555 |
| S + E + H + D | 0.337 | 0.953 | 6 | 0.287 | 103.677 | 0.848 |
| S + E + H + A | 0.397 | 0.944 | 6 | 0.311 | 87.122 | 0.843 |
| S + E + D + A | 0.422 | 0.892 | 4 | 0.419 | 68.166 | 0.843 |
| S + E + H + D + A | 0.355 | 0.944 | 6 | 0.310 | 87.636 | 0.769 |
| Compound | Predicted pIC50 | Total-Score | |
|---|---|---|---|
| CoMFA | CoMSIA | ||
| 19 | 8.788 | 8.774 | 9.17 |
| d1 | 8.903 | 9.293 | 8.62 |
| d2 | 9.393 | 8.447 | 7.20 |
| d3 | 8.360 | 8.949 | 9.02 |
| d4 | 8.547 | 8.940 | 6.53 |
| d5 | 8.998 | 9.286 | 7.27 |
| d6 | 8.726 | 9.470 | 6.57 |
| d7 | 8.603 | 9.347 | 8.36 |
| d8 | 8.871 | 9.116 | 6.68 |
| d9 | 8.833 | 8.731 | 7.13 |
| d10 | 8.552 | 8.837 | 6.50 |
| d11 | 8.730 | 9.027 | 7.82 |
| d12 | 8.628 | 9.517 | 7.51 |
| d13 | 9.082 | 8.713 | 5.89 |
| d14 | 9.094 | 9.719 | 8.45 |
| d15 | 8.722 | 9.507 | 7.30 |
| d16 | 8.527 | 9.345 | 9.25 |
| d17 | 9.115 | 8.675 | 5.99 |
 | ||||
|---|---|---|---|---|
| Compd. No. | Substituent | |||
| R1 | R2 | R3 | R4 | |
| 1 | Me | NH2 | H | H |
| 2 |  | NH2 | H | H |
| 3 |  | NH2 | H | H |
| 4 | H | NH2 | H | H |
| 5 |  | NH2 | H | H |
| 6 | i-Pr | NH2 | H | H |
| 7 |  | NH2 | H | H |
| 8 |  | NH2 | H | H |
| 9 | Me | OEt | H | H |
| 10 | Me | OH | H | H |
| 11 | Me | NHMe | H | H |
| 12 | Me | NHcyclopropyl | H | H |
| 13 | Me | NHcyclopentyl | H | H |
| 14 | Me | NHPh | H | H |
| 15 | Me | NH2 | o-CF3 | H |
| 16 | Me | NH2 | m-CF3 | H |
| 17 | Me | NH2 | p-CF3 | H |
| 18 | Me | NH2 | o-Ac | H |
| 19 | Me | NH2 | m-Ac | H |
| 20 | Me | NH2 | p-Ac | H |
| 21 | Me | NH2 | o-OMe | H |
| 22 | Me | NH2 | m-OMe | H |
| 23 | Me | NH2 | p-OMe | H |
| 24 | Me | NH2 | o-NO2 | H |
| 25 | Me | NH2 | m-NO2 | H |
| 26 | Me | NH2 | p-NO2 | H |
| 27 | Me | NH2 | o-Me | H |
| 28 | Me | NH2 | o-SMe | H |
| 29 | Me | NH2 | o-NHMe | H |
| 30 | Me | NH2 | o-F | H |
| 31 | Me | NH2 | o- i-Pr | H |
| 32 | Me | NH2 | o-CO2Me | H |
| 33 | Me | NH2 | o-CONH2 | Cl |
| 34 | Me | NH2 | o-SO2NH2 | H |
| 35 | Me | NH2 | o-Ph | H |
| 36 | Me | NH2 | o-OPh | H |
| 37 | Me | NH2 | o-benzyl | H |
| 38 | Me | NH2 | o-NHPh | H |
| 39 | Me | NH2 | o-benzoyl | H |
| 40 | Me | NH2 | o-SPh | H |
| 41 | Me | NH2 | o-NH2 | H |
| 42 | Me | NH2 | o-NHAc | H |
| 43 | Me | NH2 | o-Ac | 3′-(4-methyl-piperazin-1-yl) |
| 44 | Me | NH2 | o-Ac | 4′-(4-methyl-piperazin-1-yl) |
| 45 | Me | NH2 | o-Ac | 5′-(4-methyl-piperazin-1-yl) |
| 46 | Me | NH2 | o-OMe | 4′-(4-methyl-piperazin-1-yl) |
| 47 | Me | NH2 | o-OMe | 5′-(4-methyl-piperazin-1-yl) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ai, Y.; Wang, S.-T.; Sun, P.-H.; Song, F.-J. Molecular Modeling Studies of 4,5-Dihydro-1H-pyrazolo[4,3-h] quinazoline Derivatives as Potent CDK2/Cyclin A Inhibitors Using 3D-QSAR and Docking. Int. J. Mol. Sci. 2010, 11, 3705-3724. https://doi.org/10.3390/ijms11103705
Ai Y, Wang S-T, Sun P-H, Song F-J. Molecular Modeling Studies of 4,5-Dihydro-1H-pyrazolo[4,3-h] quinazoline Derivatives as Potent CDK2/Cyclin A Inhibitors Using 3D-QSAR and Docking. International Journal of Molecular Sciences. 2010; 11(10):3705-3724. https://doi.org/10.3390/ijms11103705
Chicago/Turabian StyleAi, Yong, Shao-Teng Wang, Ping-Hua Sun, and Fa-Jun Song. 2010. "Molecular Modeling Studies of 4,5-Dihydro-1H-pyrazolo[4,3-h] quinazoline Derivatives as Potent CDK2/Cyclin A Inhibitors Using 3D-QSAR and Docking" International Journal of Molecular Sciences 11, no. 10: 3705-3724. https://doi.org/10.3390/ijms11103705
APA StyleAi, Y., Wang, S. -T., Sun, P. -H., & Song, F. -J. (2010). Molecular Modeling Studies of 4,5-Dihydro-1H-pyrazolo[4,3-h] quinazoline Derivatives as Potent CDK2/Cyclin A Inhibitors Using 3D-QSAR and Docking. International Journal of Molecular Sciences, 11(10), 3705-3724. https://doi.org/10.3390/ijms11103705