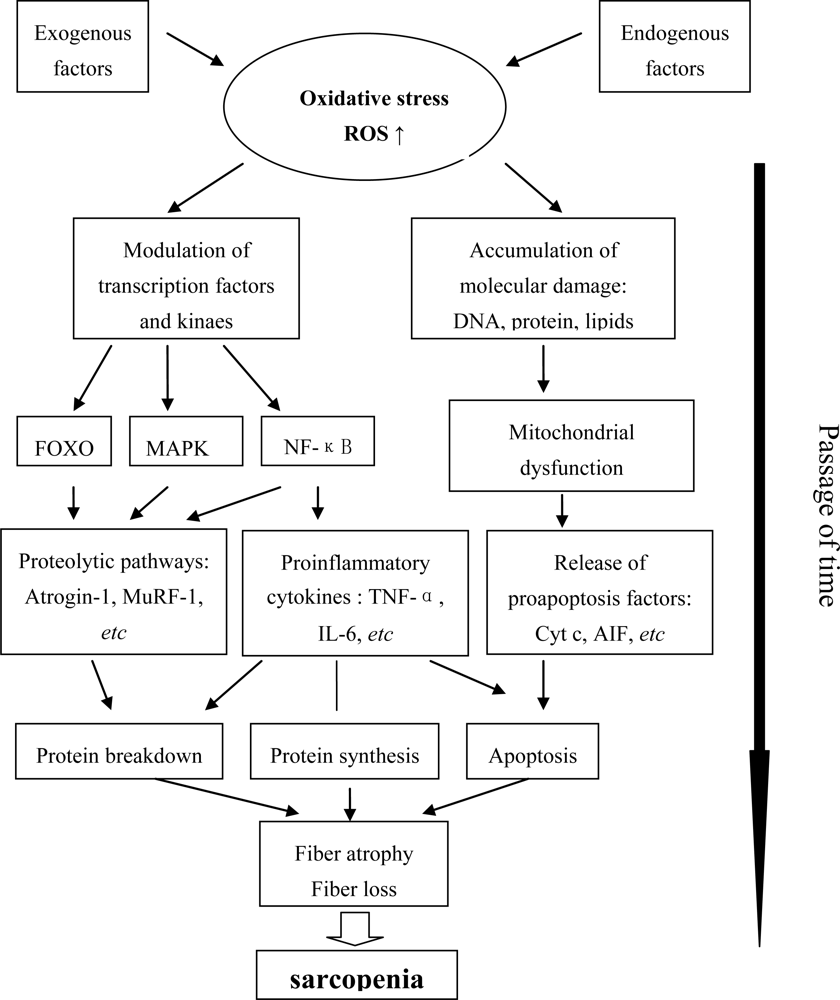
Oxidative Stress, Molecular Inflammation and Sarcopenia

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The State of Oxidative Stress and Molecular Inflammation in Aging Muscle
2.1. Oxidative Stress
2.2. Chronic Molecular Inflammation
2.3. Mitochondrial Dysfunction
3. Signaling Pathways and Kinases Involved in Age-Related Muscle Atrophy
3.1. IGF-1/Akt/mTOR
3.2. FOXO
3.3. NF-κB
3.4. MAPK
3.5. MuRF1 and Atrogin-1
3.6. PGC-1α
4. Apoptosis Signaling
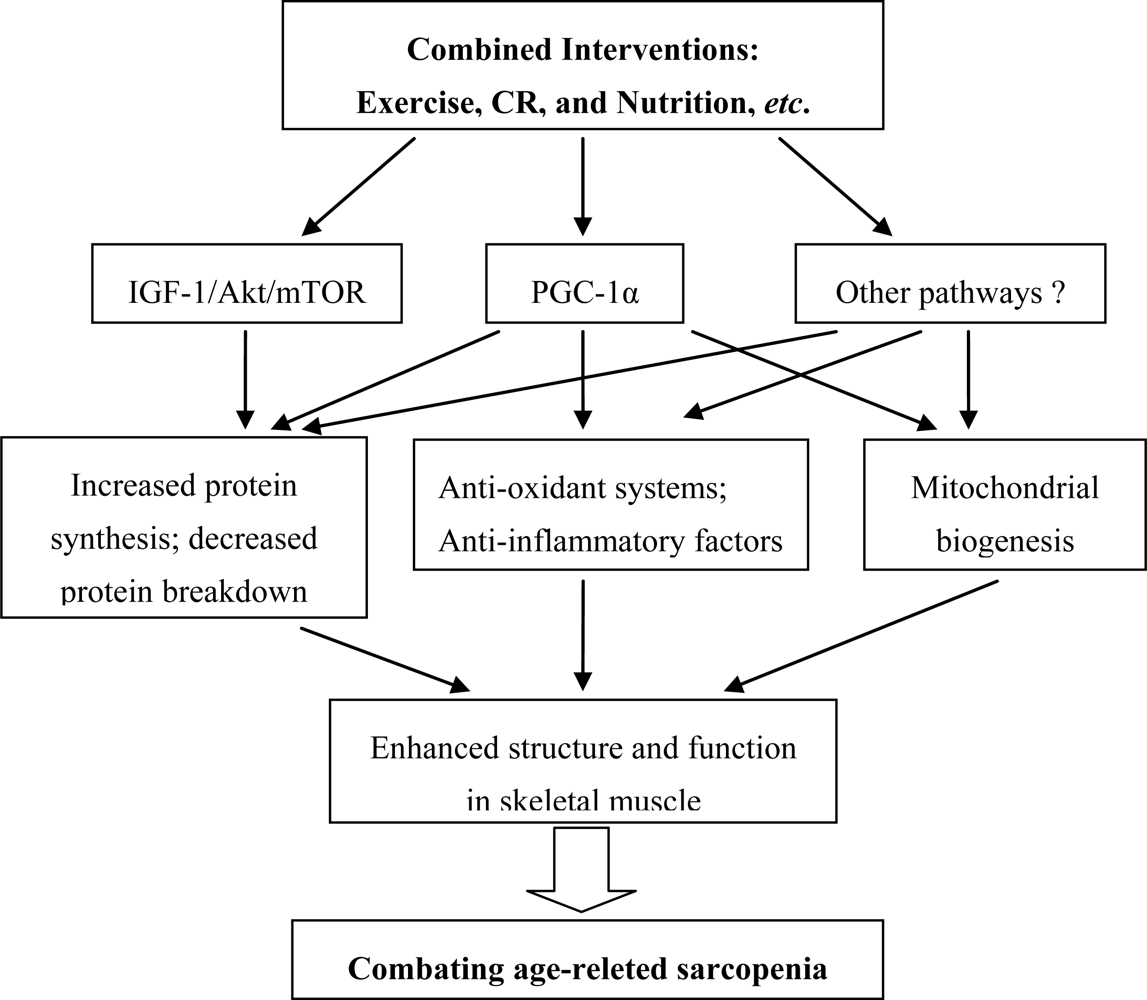
5. Considerations for Combined Interventions
5.1. Exercise
5.2. Caloric Restriction (CR)
5.3. Combined Interventions
References
- Rosenberg, IH. Sarcopenia: Origins and clinical relevance. J. Nutr 1997, 127, 990S–991S. [Google Scholar]
- Faulkner, JA; Larkin, LM; Claflin, DR; Brooks, SV. Age-related changes in the structure and function of skeletal muscles. Clin. Exp. Pharmacol. Physiol 2007, 34, 1091–1096. [Google Scholar]
- Janssen, I; Shepard, DS; Katzmarzyk, PT; Roubenoff, R. The healthcare costs of sarcopenia in the United States. J. Am. Geriatr. Soc 2004, 52, 80–85. [Google Scholar]
- Daw, CK; Starnes, JW; White, TP. Muscle atrophy and hypoplasia with aging: Impact of training and food restriction. J. Appl. Physiol 1988, 64, 2428–2432. [Google Scholar]
- Lexell, J; Taylor, CC; Sjostrom, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci 1988, 84, 275–294. [Google Scholar]
- Carter, CS; Hofer, T; Seo, AT; Leeuwenburgh, C. Molecular mechanisms of life- and health-span extension: Role of calorie restriction and exercise intervention. Appl. Physiol. Nutr. Metab 2007, 32, 954–966. [Google Scholar]
- Kregel, KC; Zhang, HJ. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol 2007, 292, R18–R36. [Google Scholar]
- Siu, PM; Pistilli, EE; Alway, SE. Age-dependent increase in oxidative stress in gastrocnemius muscle with unloading. J. Appl. Physiol 2008, 105, 1695–705. [Google Scholar]
- Moylan, JS; Reid, MB. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar]
- Samba, RD; Lauretani, F; Ferrucci, L. Carotenoids as protection against sarcopenia in older adults. Arch. Biochem. Biophys 2007, 458, 141–145. [Google Scholar]
- Howard, C; Ferrucci, L; Sun, K; Fried, LP; Walston, J; Varadhan, R; Guralnik, JM; Semba, RD. Oxidative protein damage is associated with poor grip strength among older women living in the community. J. Appl. Physiol 2007, 103, 17–20. [Google Scholar]
- Koopman, R; van Loon, LJ. Aging, exercise and muscle protein metabolism. J. Appl. Physiol 2009, 106, 2040–2048. [Google Scholar]
- Chung, HY; Cesari, M; Anton, S; Marzetti, E; Giovannini, S; Seo, AY; Carter, C; Yu, BP; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev 2009, 8, 18–30. [Google Scholar]
- Reid, MB; Li, YP. Tumor necrosis factor-α and muscle wasting: A cellular perspective. Respir. Res 2001, 2, 269–272. [Google Scholar]
- Brinkley, TE; Leng, X; Miller, ME; Kitzman, DW; Pahor, M; Berry, MJ; Marsh, AP; Kritchevsky, SB; Nicklas, BJ. Chronic inflammation is associated with low physical function in older adults across multiple comorbidities. J. Gerontol. A. Biol. Sci. Med. Sci 2009, 64, 455–461. [Google Scholar]
- Toth, MJ; Matthews, DE; Tracy, RP; Previs, MJ. Age-related differences in skeletal muscle protein synthesis: Relation to markers of immune activation. Am. J. Physiol. Endocrinol. Metab 2005, 288, E883–E891. [Google Scholar]
- Roubenoff, R. Catabolism of aging: Is it an inflammatory process? Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 295–299. [Google Scholar]
- Bua, E; Johnson, J; Herbst, A; Delong, B; McKenzie, D; Salamat, S; Aiken, JM. Mitochondrial DNA–deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet 2006, 79, 469–480. [Google Scholar]
- Mariappan, N; Elks, CM; Fink, B; Francis, J. TNF-induced mitochondrial damage: A link between mitochondrial complex I activity and left ventricular dysfunction. Free Radic Biol. Med 2009, 46, 462–470. [Google Scholar]
- Figueiredo, PA; Mota, MP; Appell, HJ; Duarte, JA. The role of mitochondria in aging of skeletal muscle. Biogerontology 2008, 9, 67–84. [Google Scholar]
- Marzetti, E; Wohlgemutz, SE; Lees, HA; Chung, H; Giovannini, S; Leeuwenburgh, C. Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mech. Ageing Dev 2008, 129, 542–549. [Google Scholar]
- Rommel, C; Bodine, SC; Clarke, BA; Rossman, R; Nunez, L; Stitt, TN; Yancopoulous, GD; Glass, DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol 2001, 3, 1009–1013. [Google Scholar]
- Sacheck, JM; Ohtsuka, A; McLary, SC; Goldberg, AL. IGF-1 stimulates muscle growth by suppressing protein breakdown and expression of atrphy-related ubiquitin-ligases, atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab 2004, 287, E591–E601. [Google Scholar]
- Li, M; Li, C; Parkhouse, WS. Age-related differences in the des IGF-I-mediated activation of Akt-1 and p70 S6K in mouse skeletal muscle. Mech. Ageing Dev 2003, 124, 771–778. [Google Scholar]
- Lai, KM; Gonzalez, M; Poueymirou, WT; Kline, WO; Na, E; Zlotchenko, E; Stitt, TN; Economides, AN; Yancopoulos, GD; Glass, DJ. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol. Cell Biol 2004, 24, 9295–9304. [Google Scholar]
- Léger, B; Cartoni, R; Praz, M; Lamon, S; Dériaz, O; Crettenand, A; Gobelet, C; Rohmer, P; Konzelmann, M; Luthi, F; et al. Akt signaling through GSK-3beta, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J. Physiol 2006, 576, 923–933. [Google Scholar]
- Latres, E; Amini, AR; Amini, AA; Griffiths, J; Martin, FJ; Wei, Y; Lin, HC; Yancopoulous, GD; Glass, DJ. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J. Biol. Chem 2005, 280, 2737–2744. [Google Scholar]
- Bodine, SC; Stitt, TN; Gonzalez, M; Kline, WO; Stover, GL; Bauerlein, R; Zlotchenko, E; Scrimgeour, A; Lawrence, JC; Glass, DJ; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nature Cell Biol 2001, 3, 1014–1019. [Google Scholar]
- Bodine, SC. mTOR signaling and the molecular adaptation to resistance exercise. Med. Sci. Sprots Exerc 2006, 38, 1950–1957. [Google Scholar]
- Koopman, R; Zorenc, AH; Gransier, RJ; Cameron-Smith, D; van Loon, LJ. Increase in S6K1 phosphorylation in human skeletal muscle following resistance exercise occurs mainly in type II muscle fibers. Am. J. Physiol. Endocrinol. Metab 2006, 290, E1245–E1252. [Google Scholar]
- Owino, V; Yang, SY; Goldspink, G. Age-related loss of skeletal muscle function and the inability to express the autocrine form of insulin-like growth factor-1 (MGF) in response to mechanical overload. FEBS Lett 2001, 505, 259–263. [Google Scholar]
- Haddad, F; Adams, GR. Aging-sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. J. Appl. Physoil 2006, 100, 1188–1203. [Google Scholar]
- O’Connor, JC; McCusker, RH; Strle, K; Johnson, RW; Dantzer, R; Kelley, KW. Regulation of IGF-I Function by Proinflammatory Cytokines: At the Interface of Immunology and Endocrinology. Cell Immunol 2008, 252, 91–110. [Google Scholar]
- Papaconstantinou, J. Insulin/IGF-1 and ROS signaling pathway cross-talk in aging and longevity determination. Mol. Cell Endocrinol 2009, 299, 89–100. [Google Scholar]
- Grounds, MD; Radley, HG; Gebski, BG; Bogoyevitch, MA; Shavlakadze, T. Implications of cross-talk between TNF and IGF-1 signalling in skeletal muscle. Proc. Aust. Physiol. Soci 2008, 39, 7–13. [Google Scholar]
- Thomson, DM; Gordon, SE. Impaired overload-induced muscle growth is associated with diminished translational signaling in aged rat fast-twitch skeletal muscle. J. Physiol 2006, 574, 291–305. [Google Scholar]
- Funai, F; Parkington, JD; Carambula, S; Fielding, RA. Age associated decrease in contraction-induced activation of downstream targets of Akt/mTOR signaling in skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol 2006, 290, R1080–R1086. [Google Scholar]
- Léger, B; Bock, KD; Hespel, P; Russell, AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of akt phosphorylation. Rejuv. Res 2008, 11, 163–175. [Google Scholar]
- Marzani, B; Balage, M; Vénien, A; Astruc, T; Papet, I; Dardevet, D; Mosoni, L. Antioxidant Supplementation Restores Defective Leucine Stimulation of Protein Synthesis in Skeletal Muscle from Old Rats. J. Nutr 2008, 138, 2205–2211. [Google Scholar]
- Furukawa-Hibi, Y; Kobayashi, Y; Chen, C; Motoyama, N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid Redox Signal 2005, 7, 752–760. [Google Scholar]
- Southgate, RJ; Neill, B; Prelovsek, O; El-Osta, A; Kamei, Y; Miura, S; Ezaki, O; McLoughlin, TJ; Zhang, W; Unterman, TG. FOXO1 regulates the expression of 4E-BP1 and inhibits mTOR signaling in mammalian skeletal muscle. J. Biol. Chem 2007, 282, 21176–21186. [Google Scholar]
- Kamei, Y; Miura, S; Suzuki, M; Kai, Y; Mizukami, J; Taniguchi, T; Mochida, K; Hata, T; Matsuda, J; Aburatani, H; et al. Skeletal muscle FOXO1(FKHR) transgenic mice have less skeletal muscle mass, down-regulated type I (slow twitch /red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem 2004, 279, 41114–41123. [Google Scholar]
- Stitt, TN; Drujan, D; Clarke, BA; Panaro, F; Timofeyva, Y; Kline, WO; Gonzalez, M; Yancopoulos, GD; Glass, DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar]
- Welle, S; Brooks, AI; Delehanty, JM; Needler, N; Thornton, CA. Gene expression profile of aging in human muscle. Physiol. Genomics 2003, 14, 149–159. [Google Scholar]
- Machida, S; Booth, FW. Increased nuclear proteins in muscle satellite cells in aged animals as compared to young growing animals. Exp. Gerontol 2004, 39, 1521–1525. [Google Scholar]
- Sandri, M; Sandri, C; Gilbert, A; Skurk, C; Calabria, E; Picard, A; Walsh, K; Schiaffino, S; Lecker, SH; Goldberg, AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar]
- Giresi, PG; Stevenson, EJ; Theilhaber, J; Koncarevic, A; Parkington, J; Fielding, RA; Kandarian, SC. Identification of a molecular signature of sarcopenia. Physiol. Genomics 2005, 21, 253–263. [Google Scholar]
- Piette, J; Piret, B; Bonizzi, G; Schoonbroodt, S; Merville, MP; Legrand-Poels, S; Bours, V. Multiple redox regulation in NF-κB transcription factor activation. Biol Chem 1997, 378. [Google Scholar]
- Dogra, C; Changotra, H; Mohan, S; Kumar, A. Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J. Biol. Chem 2006, 281, 10327–10336. [Google Scholar]
- Mourkioti, F; Kratsios, P; Luedde, T; Song, Y; Delafontaine, P; Adami, R; Parente, V; Bottinelli, R; Pasparakis, M; Rosenthal, N. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Invest 2006, 116, 2945–2954. [Google Scholar]
- Hunter, RB; Kandarian, SC. Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J. Clin. Invest 2004, 114, 1504–1511. [Google Scholar]
- Urso, ML; Chen, YW; Scrimgeour, AG; Lee, PC; Lee, KF; Clarkson, PM. Alterations in mRNA expression and protein products following spinal cord injury in humans. J. Physiol 2007, 579, 877–892. [Google Scholar]
- Cuthbertson, D; Smith, K; Babraj, J; Leese, G; Waddell, T; Atherton, P; Wackerhage, H; Taylor, PM; Rennie, MJ. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J 2005, 19, 422–424. [Google Scholar]
- Phillips, T; Leeuwenburgh, C. Muscle fiber specific apoptosis and TNF-α signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J 2005, 19, 668–670. [Google Scholar]
- Li, YP; Chen, Y; John, J; Moylan, J; Jin, B; Mann, DL; Reid, MB. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 2005, 19, 362–370. [Google Scholar]
- Gredinger, E; Gerber, A; Tamir, Y; Tapscott, S; Bengal, E. Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J. Biol. Chem 1998, 273, 10436–10444. [Google Scholar]
- Musaro, A; De Angelis, C; Germani, A; Ciccarelli, C; Molinaro, M; Zani, B. Enhanced expression of myogenic regulatory genes in aging skeletal muscle. Exp. Cell Res 1995, 221, 241–248. [Google Scholar]
- Williamson, D; Gallagher, P; Harber, M; Hollon, C; Trappe, S. Mitogen-activated protein kinase (MAPK) pathway activation: Effects of age and acute exercise on human skeletal muscle. J. Physiol 2003, 547, 977–987. [Google Scholar]
- Drummond, MJ; Dreyer, HC; Pennings, B; Fry, CS; Dhanani, S; Dillon, EL; Sheffield-Moore, M; Volpi, E; Rasmussen, BB. Skeletal muscle protein anabolic response to resistance exercise and essential amino acids is delayed with aging. J. Appl. Physiol 2008, 104, 1452–1461. [Google Scholar]
- Raingeaud, J; Gupta, S; Rogers, J; Dickens, M; Han, J; Ulevitch, RJ; Davis, RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem 1995, 270, 7420–7426. [Google Scholar]
- Shen, HM; Liu, ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic. Biol. Med 2006, 40, 928–939. [Google Scholar]
- Braga, M; Sinha-Hikim, AP; Datta, S; Ferrini, MG; Brown, D; Kovacheva, EL; Gonzalez-Cadavid, NF; Sinha-Hikim, I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis 2008, 13, 822–832. [Google Scholar]
- Glass, DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol 2005, 37, 1974–1984. [Google Scholar]
- Bodine, SC; Latres, E; Baumhueter, S; Lai, VK; Nunez, L; Clarke, BA; Poueymirou, WT; Panaro, FJ; Na, E; Dharmarajan, K; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar]
- Li, HH; Kedar, V; Zhang, C; Arya, R; Wang, DZ; Patterson, C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Invest 2004, 114, 1058–1071. [Google Scholar]
- Stitt, TN; Drujan, D; Clarke, BA; Panaro, FJ; Timofeyva, Y; Kline, WO; Gonzalez, M; Yancopoulos, GD; Glass, DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar]
- Cai, D; Frantz, JD; Tawa, NE, Jr; Melendez, PA; Oh, B; Lidov, HGW; Hasselgren, P; Frontera, WR; Lee, J; Glass, DJ; et al. IKKb/NF-kB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar]
- Clavel, S; Coldefy, AS; Kurkdjian, E; Salles, J; Margaritis, I; Derijard, B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat tibialis anterior muscle. Mech. Ageing Dev 2006, 127, 794–801. [Google Scholar]
- Edstrom, E; Altun, M; Hagglund, M; Ulfhake, B. Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. J. Gerontol. A: Biol. Sci. Med. Sci 2006, 61, 663–674. [Google Scholar]
- Whitman, SA; Wacker, MJ; Richmond, SR; Godard, MP. Contributions of the ubiquitin-proteasome pathway and apoptosis to human skeletal muscle wasting with age. Pflugers Arch 2005, 450, 437–446. [Google Scholar]
- Raue, U; Slivka, D; Jemiolo, B; Hollon, C; Trappe, S. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J. Gerontol. A Biol. Sci. Med. Sci 2007, 62, 1407–1412. [Google Scholar]
- Christoph, H; Spiegelman, M. The role of exercise and PGC1α in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar]
- Ventura-Clapier, R; Garnier, A; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1α. Cardiovasc. Res 2008, 79, 208–217. [Google Scholar]
- Sandri, M; Lin, J; Handschin, C; Yang, W; Arany, ZP; Lecker, SH; Goldberg, AL; Spiegelman, BM. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar]
- Handschin, C; Kobayashi, YM; Chin, S; Seale, P; Campbell, KP; Spiegelman, BM. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 2007, 21, 770–783. [Google Scholar]
- St-Pierre, J; Drori, S; Uldry, M; Silvaggi, JM; Rhee, J; Jäger, S; Handschin, C; Zheng, K; Lin, K; Yang, W; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar]
- Hood, DA; Irrcher, I; Ljubicic, V; Joseph, AM. Coordination of metabolic plasticity in skeletal muscle. J. Exp. Bio 2006, 209, 2265–2275. [Google Scholar]
- Jager, S; Handschin, C; St-Pierre, J; Spiegelman, BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar]
- Chabi, B; Ljubicic, V; Menzies, KJ; Huang, JH; Sallem, A; Hood, DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar]
- López-Lluch, G; Irusta, PM; Navas, P; de Cabo, R. Mitochondrial biogenesis and healthy aging. Exp. Gerontol 2008, 43, 813–819. [Google Scholar]
- Leeuwenburgh, C. Role of apoptosis in sarcopenia. J. Gerontol. Med. Sci 2003, 58, 999–1001. [Google Scholar]
- Nitahara, JA; Cheng, W; Liu, Y; Li, B; Leri, A; Mogul, D; Gambert, SR; Kajstura, J; Anversa, P. Intracellular calcium, DNase activity and myocyte apoptosis in aging Fischer 344 rats. J. Mol. Cell. Cardiol 1998, 30, 519–535. [Google Scholar]
- Cadenas, E; Davies, KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Rad. Biol. Med 2000, 29, 222–230. [Google Scholar]
- Song, W; Kwak, H; Lawler, JM. Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxid. Redox. Signal 2006, 8, 517–528. [Google Scholar]
- Greiwe, JS; Cheng, B; Rubin, DC; Yarasheski, KE; Semenkovich, CF. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 2001, 15, 475–482. [Google Scholar]
- Menshikova, EV; Ritov, VB; Fairfull, L; Ferrell, RE; Kelley, DE; Goodpaster, BH. Effects of Exercise on Mitochondrial Content and Function in Aging Human Skeletal Muscle. J. Gerontol. A: Biol. Sci. Med. Sci 2006, 61, 534–540. [Google Scholar]
- Russell, AP; Schreiber, S; Crettenand, A; Meier, CA; Kralli, A; Deriaz, O. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 2003, 52, 2874–2881. [Google Scholar]
- Gleeson, M; McFarlin, B; Flynn, M. Exercise and Toll-like receptors. Exerc. Immunol. Rev 2006, 12, 34–53. [Google Scholar]
- Petersen, AM; Pedersen, BK. The anti-inflammatory effect of exercise. J. Appl. Physiol 2005, 98, 1154–1162. [Google Scholar]
- Hameed, M; Orrell, RW; Cobbold, M; Goldspink, G; Harridge, SD. Expression of IGF-I splice variants in young and old human skeletal muscle after high resistance exercise. J. Physiol 2003, 547, 247–254. [Google Scholar]
- Kosek, DJ; Kim, J; Petrella, JK; Cross, JM; Bamman, MM. Efficacy of 3 days/wk resistance training on myofiber hypertrophy and myogenic mechanisms in young vs. older adults. J. Appl. Physiol 2006, 101, 531–544. [Google Scholar]
- Melov, S; Tarnopolsky, MA; Beckman, K; Felkey, K; Hubbard, A. Resistance exercise reverses aging in human skeletal muscle. PLoS ONE 2007, 2, e465. [Google Scholar]
- Sillanpää, E; Häkkinen, A; Nyman, K; Mattila, M; Cheng, S; Karavirta, L; Laaksonen, DE; Huuhka, N; Kraemer, WJ; Häkkinen, K. Body Composition and Fitness during Strength and/or Endurance Training in Older Men. Med. Sci. Sports Exerc 2008, 40, 950–958. [Google Scholar]
- Nader, GA. Concurrent strength and endurance training: From molecules to man. Med. Sci. Sports Exerc 2006, 38, 1965–1970. [Google Scholar]
- Atherton, PJ; Babraj, J; Smith, K; Singh, J; Rennie, MJ; Wackerhage, H. Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J 2005, 19, 786–798. [Google Scholar]
- Leeuwenburgh, C; Wagner, P; Holloszy, JO; Sohal, RS; Heinecke, JW. Caloric restriction attenuates dityrosine cross-linking of cardiac and skeletal muscle proteins in aging mice. Arch. Biochem. Biophys 1997, 346, 74–80. [Google Scholar]
- McKiernan, S; Bua, E; McGorray, J; Aiken, J. Early-onset calorie restriction conserves fiber number in aging rat skeletal muscle. FASEB J 2004, 18, 580–581. [Google Scholar]
- Kim, J; Kwak, H; Leeuwenburgh, C; Lawler, JM. Lifelong Exercise and Mild (8%) Caloric Restriction Attenuate Age-induced Alterations in Plantaris Muscle Morphology, Oxidative Stress and IGF-1 in the Fischer-344 Rat. Exp. Gerontol 2008, 43, 317–329. [Google Scholar]
- Dreyer, HC; Volpi, E. Role of protein and amino acids in the pathophysiology and treatment of sarcopenia. J. Am. Coll. Nutr 2005, 24, 140S–145S. [Google Scholar]
- Fujita, S; Dreyer, HC; Drummond, MJ; Glynn, EL; Cadenas, JG; Yoshizawa, F; Volpi, E; Rasmussen, BB. Nutrient signalling in the regulation of human muscle protein synthesis. J. Physiol 2007, 582, 813–823. [Google Scholar]
- Karlsson, HK; Nilsson, PA; Nilsson, J; Chibalin, AV; Zierath, JR; Blomstrand, E. Branched-chain amino acids increase p70S6k phosphorylation in human skeletal muscle after resistance exercise. Am. J. Physiol 2004, 287, E1–E7. [Google Scholar]
- Zangarelli, A; Chanseaume, E; Morio, B; Brugère, C; Mosoni, L; Rousset, P; Giraudet, C; Patrac, V; Gachon, P; Boirie, Y; et al. Synergistic effects of caloric restriction with maintained protein intake on skeletal muscle performance in 21-month-old rats: A mitochondria-mediated pathway. FASEB J 2006, 20, 2439–2450. [Google Scholar]


© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Meng, S.-J.; Yu, L.-J. Oxidative Stress, Molecular Inflammation and Sarcopenia. Int. J. Mol. Sci. 2010, 11, 1509-1526. https://doi.org/10.3390/ijms11041509
Meng S-J, Yu L-J. Oxidative Stress, Molecular Inflammation and Sarcopenia. International Journal of Molecular Sciences. 2010; 11(4):1509-1526. https://doi.org/10.3390/ijms11041509
Chicago/Turabian StyleMeng, Si-Jin, and Long-Jiang Yu. 2010. "Oxidative Stress, Molecular Inflammation and Sarcopenia" International Journal of Molecular Sciences 11, no. 4: 1509-1526. https://doi.org/10.3390/ijms11041509
APA StyleMeng, S. -J., & Yu, L. -J. (2010). Oxidative Stress, Molecular Inflammation and Sarcopenia. International Journal of Molecular Sciences, 11(4), 1509-1526. https://doi.org/10.3390/ijms11041509