Diversity of Phylogenetic Information According to the Locus and the Taxonomic Level: An Example from a Parasitic Mesostigmatid Mite Genus

Abstract
:1. Introduction
- Address the following questions about the evolution of host specificity in Dermanyssus:
- Are the lineages of the generalist D. gallinae’s lineages effectively composed of cryptic species (potentially making them as specialized as any of the four specialist species)?
- Is the generalist condition derived or ancestral?
2. Material and Methods
2.1. Biological Material
2.2. DNA Data
2.3. Datasets
Step 1: multi-gene analyses
Step 2: multi-isolate analyses
2.4. Phylogenetics
2.4.1. Phylogenetic Analyses
2.4.2. Outgroups
2.4.3. Clade Robustness Support Values
2.4.4. Comparison of Mitochondrial versus Nuclear Monophylies
2.5. Statistical Analysis of Haplotype Frequencies and Diversity
3. Results
3.1. DNA Sequences
3.1.1. Alignments of Obtained Gene Fragments
3.1.2. Molecular Characteristics of Obtained Gene Fragments
3.2. Step 1: Multi-Gene Analyses
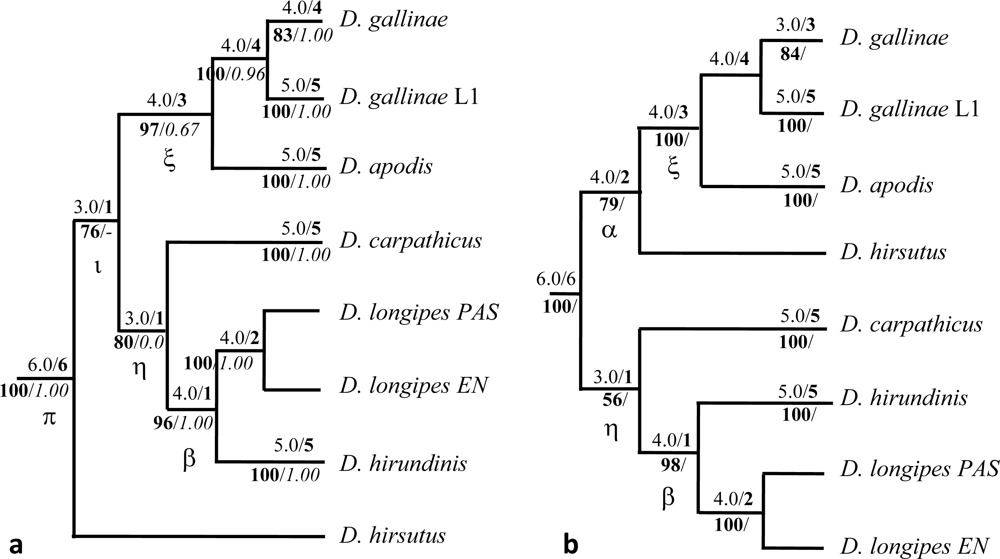
3.2.1. Phylogenetic Interrelationships at the Specific Level
3.2.2. Specific Characterization Power of Sequences
- COI, 16S and Tropomyosin intron n are very informative at the specific and intraspecific levels, as usually noted in other arthropods.
- 5.8S and Tropomyosin exon n and n + 1 are insufficiently informative at the specific level and do not show any intraspecific variation, as expected.
3.3. Step 2: Multi-Isolate Analyses
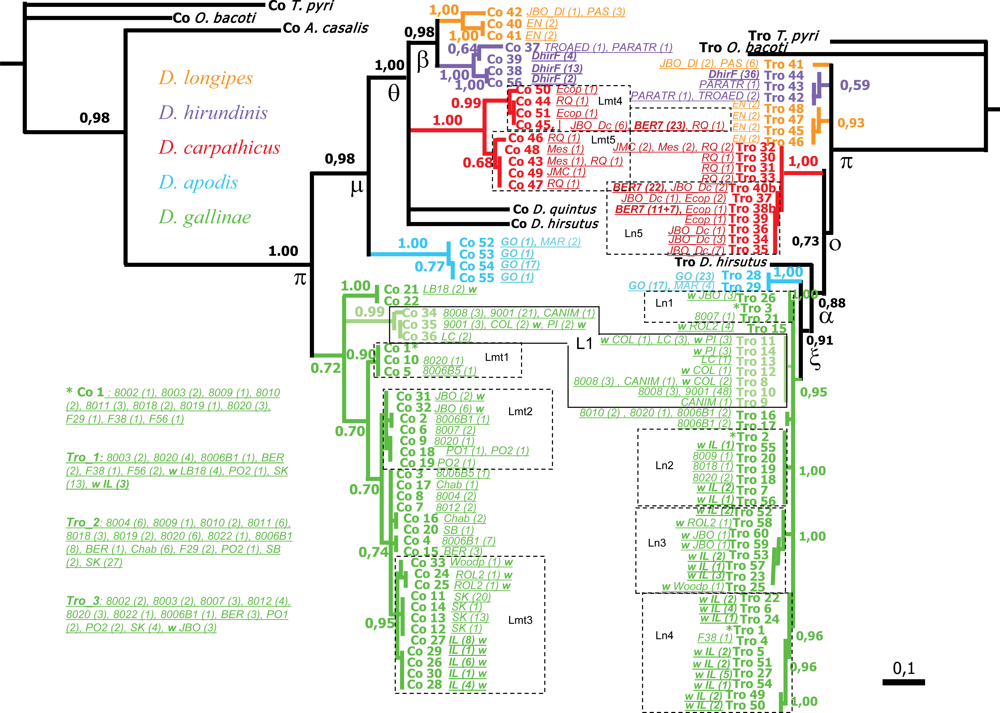
3.3.1. Additive Information about Phylogenetic Interrelationships
3.3.2. Differentiation within Previously Delimited Specific Entities
4. Discussion
4.1. Specific Characterization Power of Sequences
4.2. Phylogenetic Interrelationships at the Specific Level
4.3. Generalist: A Derived or Ancestral Condition?
4.4. Reticulate Evolution or Gradual Specialization/Speciation?
5. Conclusion
List of Electronic Supplementary Information (ESI)
ISOL_TRO1 (nexus DNA alignment of Tropomyosin partial exon n, complete intron n, partial exon n + 1 upon which the matrix of encoded indels has been established – see Appendix z3).Appendix 1 (Sampling and EMBL information for the populations under test in present study)
Appendix 6 (MP topologies obtained in Step 2 not shown in Figure 4)



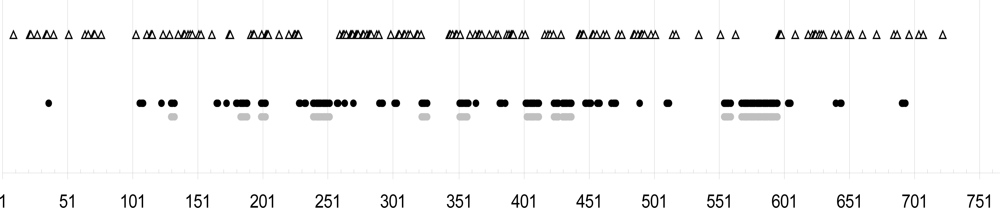
 ,
,  ,
,  ,
,  (names in bold). Within
(names in bold). Within  , a w indicates samples from wild avifauna (other species only from the wild avifauna). Note: here Bayesian topologies are displayed aiming at showing branch length, but one must keep in mind that they resulted in interrelationships similar to MP analyses. Observed host ranges: Passeriformes only:
, a w indicates samples from wild avifauna (other species only from the wild avifauna). Note: here Bayesian topologies are displayed aiming at showing branch length, but one must keep in mind that they resulted in interrelationships similar to MP analyses. Observed host ranges: Passeriformes only:  (2 Simple Isolates) on sparrows (Passeridae: Passer spp.),
(2 Simple Isolates) on sparrows (Passeridae: Passer spp.),  (1 SI) on tits (Paridae: Parus spp.),
(1 SI) on tits (Paridae: Parus spp.),  French lineage (6 SI) on swallows (Hirundinidae),
French lineage (6 SI) on swallows (Hirundinidae),  (5 SI) on redstarts (Muscicapidae: Phoenicurus sp.) and tits,
(5 SI) on redstarts (Muscicapidae: Phoenicurus sp.) and tits,  (1 Focused Isolate, 2 SI) on swifts (Apodidae: Apus sp.); Columbiformes:
(1 Focused Isolate, 2 SI) on swifts (Apodidae: Apus sp.); Columbiformes:  (1 FI, 6 SI) on pigeons (except 2 dead individuals respectively from a swift and an owl nests); Various bird orders:
(1 FI, 6 SI) on pigeons (except 2 dead individuals respectively from a swift and an owl nests); Various bird orders:  on Coraciiformes, Passeriformes, Galliformes, Apodiformes of which L3 (1 FI) on starlings (Sturnidae: Sturnus vulgaris).
, , , (names in bold). Within , a w indicates samples from wild avifauna (other species only from the wild avifauna). Note: here Bayesian topologies are displayed aiming at showing branch length, but one must keep in mind that they resulted in interrelationships similar to MP analyses. Observed host ranges: Passeriformes only: (2 Simple Isolates) on sparrows (Passeridae: Passer spp.), (1 SI) on tits (Paridae: Parus spp.), French lineage (6 SI) on swallows (Hirundinidae), (5 SI) on redstarts (Muscicapidae: Phoenicurus sp.) and tits, (1 Focused Isolate, 2 SI) on swifts (Apodidae: Apus sp.); Columbiformes: (1 FI, 6 SI) on pigeons (except 2 dead individuals respectively from a swift and an owl nests); Various bird orders: on Coraciiformes, Passeriformes, Galliformes, Apodiformes of which L3 (1 FI) on starlings (Sturnidae: Sturnus vulgaris).
on Coraciiformes, Passeriformes, Galliformes, Apodiformes of which L3 (1 FI) on starlings (Sturnidae: Sturnus vulgaris).
, , , (names in bold). Within , a w indicates samples from wild avifauna (other species only from the wild avifauna). Note: here Bayesian topologies are displayed aiming at showing branch length, but one must keep in mind that they resulted in interrelationships similar to MP analyses. Observed host ranges: Passeriformes only: (2 Simple Isolates) on sparrows (Passeridae: Passer spp.), (1 SI) on tits (Paridae: Parus spp.), French lineage (6 SI) on swallows (Hirundinidae), (5 SI) on redstarts (Muscicapidae: Phoenicurus sp.) and tits, (1 Focused Isolate, 2 SI) on swifts (Apodidae: Apus sp.); Columbiformes: (1 FI, 6 SI) on pigeons (except 2 dead individuals respectively from a swift and an owl nests); Various bird orders: on Coraciiformes, Passeriformes, Galliformes, Apodiformes of which L3 (1 FI) on starlings (Sturnidae: Sturnus vulgaris).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mtDNA | nDNA | |
|---|---|---|
| rRNA | 1comb2 (16S) | 1comb3 (5.8S) |
| Internal Transcribed Spacers | 1comb4 (ITS1 & ITS2) | |
| Protein coding genes | 1comb1 (COI) | 1comb6 (Tropomyosin exon n & n + 1, EF-1α) |
| Intron | 1comb5 (Tropomyosin intron n) |
| Isolate names | multi-gene MP analyses | multi-gene MP COI analysis | multi-gene BA COI analysis | multi-gene MP Tropomyosin analysis | multi-gene BA Tropomyosin analysis | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clade | COL,COL2,LC,LC2,PI | Chab, Chab2, LB18, PO2, ROL1, ROL2, SK, SK2, SK3, Woodp | MAR,GO | JBO59, 5, RQ, RQ2 | Adhirun, HR, OC | PAS | ENVL08_3 | ADhirs | Tropilaelaps | JBO_Os,FS5,FS6,OSBM | OB | Gaps as missing data | Gaps as the fifth state | Gaps as missing data | Gaps as the fifth state | Gaps alone | Gaps as missing data | ||||
| max. no of occurrence per partitioning scheme | Final repetition index | max. no of occurrence per partitioning scheme | Final repetition index | presence/absence of clades | presence/absence of clades | presence/absence of clades | presence/absence of clades | presence/absence of clades | presence/absence of clades | ||||||||||||
| D. gallinae | x | x | 4 | 4 | 4 | 4 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| D. gallinae L1 | x | 5 | 5 | 5 | 5 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||
| D. gallinae non L1 | x | 4 | 4 | 3 | 3 | 1 | 1 | 0 | 0 | 0 | 0 | ||||||||||
| D. apodis | x | 5 | 5 | 5 | 5 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||
| D. hirundinis | x | 5 | 5 | 5 | 5 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||
| D. carpathicus | x | 5 | 5 | 5 | 5 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||||
| D. longipes | x | x | 4 | 2 | 4 | 3 | 1 | 1 | 0 | 0 | 0 | 0 | |||||||||
| O. sylviarum | x | 5 | 5 | 5 | 5 | - | - | - | - | - | - | ||||||||||
| x | x | x | x | 4 | 3 | 4 | 3 | 0 | 0 | 1 | 1 | 1 | 1 | ||||||||
| b | x | x | x | 4 | 1 | 4 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||
| g | x | x | 2 | −2 | 2 | −2 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||||
| d | x | x | 3 | −1 | 3 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||||
| h | x | x | x | x | 3 | 1 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |||||||
| q | x | x | x | x | x | 1 | −2 | 0 | − | 1 | 1 | 0 | 0 | 0 | 0 | ||||||
| a | x | x | x | x | 0 | - | 4 | 2 | 0 | 0 | 1 | 1 | 1 | 1 | |||||||
| m | x | x | x | x | x | x | 1 | −1 | 2 | −2 | 0 | 0 | 0 | 0 | 0 | 0 | |||||
| i | x | x | x | x | x | x | x | 3 | 1 | 2 | −2 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| n | x | x | x | x | x | x | x | 2 | −1 | 2 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| y | x | x | x | x | x | x | x | 1 | −3 | 1 | −3 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| z | x | x | x | x | x | x | 0 | - | 0 | - | 1 | 1 | 0 | 0 | 0 | 0 | |||||
| o | x | x | x | x | x | x | x | 0 | - | 1 | −1 | 0 | 0 | 0 | 1 | 1 | 0 | ||||
| p | x | x | x | x | x | x | x | x | 6 | 6 | 6 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| r | x | x | x | x | x | x | x | x | x | 2 | 2 | 0 | - | 0 | 0 | 0 | 0 | 0 | 0 | ||
| s | x | x | 6 | 6 | 5 | 5 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||
| f | x | x | x | x | x | 0 | - | 0 | - | 0 | 0 | 0 | 1 | 0 | 1 | ||||||
| m | x | x | x | x | x | x | 0 | - | 0 | - | 1 | 1 | 0 | 0 | 0 | 0 | |||||
| Between species | Within species | Between D. gallinae L1 and D. gallinae non L1 | Between D. longipes EN and D. longipes PAS | Remarks | |
|---|---|---|---|---|---|
| COI | 9–18% | 0–5% (rarely up to 9%) | 10–12% | 5% | |
| 16 S | 10–16% | 0–4% | 6–7% | 3% | |
| 5.8 S | 0–3% | 0% | 0% | 0% | only D. carpathicus and D. hirsutus differenciated from each other and from others. |
| ITS1 and 2 | 2–5% (rarely up to 9%) | 1% | 3% | 2% | 9% between D. hirsutus and other Dermanyssus species only - More than a half: 2–3% – 1% in case between D. apodis and D. gallinae non L1, and between D. hirundinis and D. longipes EN |
| Tropomyosin intron n | 8–20% | 0–6% | 2–6% | 4% | |
| Tropomyosin exon n and n + 1 | cf. remarks | 0% | 0% | 0% | Very small portion (25 pb). 2 point mutations in D. apodis vs. other Dermanyssus species. 1–2 point mutations + 1 indel Ornithonyssus vs. Dermanyssus |
| Tropomyosin | COI | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolate/population | n | G | HET(observed) | Allind | D% ind | D% no in | S(observed) | Sind (observed) | h | h (gap as a fifth state) | Hd | K (gap as missing data) | K (gap as a fifth state) | n | S | h | Hd | K |
| D. gallinae SK | 44 | 4 | 0.41 | P | 0.03 | 0.02 | 14 | 41 | 3 | 3 | 0.54 | 4.60 | 14.46 | 24 | 3 | 4 | 0.31 | 0.33 |
| D. gallinae IL | 38 | 18 | 0.88 | P | 0.06 | 0.03 | 37 | 116 | 18 | 18 | 0.94 | 9.56 | 31.88 | 20 | 4 | 5 | 0.74 | 1.06 |
| D. gallinae L1 9001 | 48 | 1 | - | - | 0.00 | 0.00 | 0 | 0 | 1 | 1 | 0.00 | 0.00 | 0.00 | 24 | 8 | 2 | 0.23 | 1.83 |
| D. apodis GO | 40 | 3 | 0.14 | P | 0.01 | 0.01 | 4 | 9 | 2 | 2 | 0.50 | 2.01 | 4.51 | 20 | 3 | 4 | 0.28 | 0.39 |
| D. carpathicus BER7 | 40 | 5 | 0.70 | A | 0.01 | 0.01 | 4 | 4 | 3 | 4 | 0.61 | 1.83 | 1.83 | 24 | 0 | 1 | 0.00 | 0 |
| D. hirundinis DhirF | 36 | 1 | - | - | 0.00 | 0.00 | 0 | 0 | 1 | 1 | 0.00 | 0.00 | 0.00 | 21 | 15 | 4 | 0.53 | 4.59 |
| D. gallinae SK | 44 | 4 | 0.41 | P | 0.03 | 0.02 | 14 | 41 | 3 | 3 | 0.54 | 4.60 | 14.46 | 24 | 3 | 4 | 0.31 | 0.33 |
| D. gallinae IL | 38 | 18 | 0.88 | P | 0.06 | 0.03 | 37 | 116 | 18 | 18 | 0.94 | 9.56 | 31.88 | 20 | 4 | 5 | 0.74 | 1.06 |
| D. gallinae L1 9001 | 48 | 1 | - | - | 0.00 | 0.00 | 0 | 0 | 1 | 1 | 0.00 | 0.00 | 0.00 | 24 | 8 | 2 | 0.23 | 1.83 |
| D. apodis GO | 40 | 3 | 0.14 | P | 0.01 | 0.01 | 4 | 9 | 2 | 2 | 0.50 | 2.01 | 4.51 | 20 | 3 | 4 | 0.28 | 0.39 |
| D. carpathicus BER7 | 40 | 5 | 0.70 | A | 0.01 | 0.01 | 4 | 4 | 3 | 4 | 0.61 | 1.83 | 1.83 | 24 | 0 | 1 | 0.00 | 0 |
| D. hirundinis DhirF | 36 | 1 | - | - | 0.00 | 0.00 | 0 | 0 | 1 | 1 | 0.00 | 0.00 | 0.00 | 21 | 15 | 4 | 0.53 | 4.59 |
| (a) | Ratio external/internal mt branch length | Number of haplotypes | Number of isolates | Number of occurrences (COI haploid, Tpm diploid) | Bootstrap (MP gap 5th state) | Relative Bremer index (MP gap 5th state) | BPP | |
|---|---|---|---|---|---|---|---|---|
| COI | D. gallinae L1 | 7.3 | 3 | 6 | 34 | 100 | 100 | 0.99 |
| D. gallinae Lmt1 | 7.5 | 3 | 12 | 20 | 99 | 100 | 0.9 | |
| D. gallinae Lmt2 | 0.3 | 7 | 6 | 15 | 61 | 100 | 0.69 | |
| D. gallinae Lmt3 | 0.3 | 12 | 4 | 58 | 23 | 50 | 0.95 | |
| D. carpathicus Lmt4 | 4.5 | 4 | 4 | 33 | 100 | 100 | 0.99 | |
| D. carpathicus Lmt5 | 1.0 | 5 | 3 | 6 | 99 | 100 | 0.68 | |
| Tpm | D. gallinae L1 | 7 | 6 | 70 | 96 | 92 | 0.97 | |
| D. gallinae Ln1 | 3 | 13 | 38 | 100 | 88 | 1 | ||
| D. gallinae Ln2 | 7 | 16 | 81 | 97 | 85 | 1 | ||
| D. gallinae Ln3 | 8 | 4 | 12 | 92 | 94 | 1 | ||
| D. gallinae Ln4 | 11 | 11 | 55 | 81 | 83 | 0.96 | ||
| D. carpathicus Ln5 | 7 | 3 | 58 | 68 | 100 | 0.79 | ||
| COI | D. gallinae L1 | 7.3 | 3 | 6 | 34 | 100 | 100 | 0.99 |
| D. gallinae Lmt1 | 7.5 | 3 | 12 | 20 | 99 | 100 | 0.9 | |
| D. gallinae Lmt2 | 0.3 | 7 | 6 | 15 | 61 | 100 | 0.69 | |
| D. gallinae Lmt3 | 0.3 | 12 | 4 | 58 | 23 | 50 | 0.95 | |
| D. carpathicus Lmt4 | 4.5 | 4 | 4 | 33 | 100 | 100 | 0.99 | |
| D. carpathicus Lmt5 | 1.0 | 5 | 3 | 6 | 99 | 100 | 0.68 | |
| Tpm | D. gallinae L1 | 7 | 6 | 70 | 96 | 92 | 0.97 | |
| D. gallinae Ln1 | 3 | 13 | 38 | 100 | 88 | 1 | ||
| D. gallinae Ln2 | 7 | 16 | 81 | 97 | 85 | 1 | ||
| D. gallinae Ln3 | 8 | 4 | 12 | 92 | 94 | 1 | ||
| D. gallinae Ln4 | 11 | 11 | 55 | 81 | 83 | 0.96 | ||
| D. carpathicus Ln5 | 7 | 3 | 58 | 68 | 100 | 0.79 | ||
| (b) | ||||||
| L1 | Ln1 | Ln2 | Ln3 | Ln4 | Ln5 | |
| L1 | 100/100 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| Lmt1 | 0/0 | 25/23 | 58/44 | 0/0 | 33/36 | 0/0 |
| Lmt2 | 0/0 | 100/46 | 50/19 | 17/17 | 50/27 | 0/0 |
| Lmt3 | 0/0 | 50/15 | 50/13 | 75/75 | 50/18 | 0/0 |
| Lmt4 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 75/100 |
| Lmt5 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| gapmode | No of trees | Tree length | CI | CI excluding uninformative characters | RI | ||
|---|---|---|---|---|---|---|---|
| multi-gene analyses | 6comb | missing | 1 | 1911 | 0.6787 | 0.6375 | 0.8529 |
| 6comb | 5th state | 1 | 2685 | 0.6685 | 0.6421 | 0.8623 | |
| COI (1comb1) | - | 7 | 738 | 0.5068 | 0.4793 | 0.7776 | |
| 16S (1comb2) | missing | 5 | 296 | 0.6926 | 0.6527 | 0.8551 | |
| 16S (1comb2) | 5th state | 5 | 352 | 0.7131 | 0.6863 | 0.8758 | |
| 5.8S (1comb3) | missing | >1000 | 48 | 0.875 | 0.8235 | 0.9155 | |
| ITS1_2 (1comb4) | missing | 4 | 283 | 0.8375 | 0.784 | 0.891 | |
| ITS1_2 (1comb4) | 5th state | 8 | 376 | 0.8457 | 0.7986 | 0.8854 | |
| TropoINTR (1comb5) | missing | 18 | 492 | 0.8882 | 0.8721 | 0.9608 | |
| TropoINTR (1comb5) | 5th state | 126 | 911 | 0.8804 | 0.8685 | 0.9635 | |
| TropoEX (1comb6) | - | 1 | 3 | 1 | 1 | ||
| EF-1α | - | 140 | 165 | 0.8848 | 0.7738 | 0.8545 | |
| multi-isolate analyses | Tropo | missing | 352 | 726 | 0.8278 | 0.7845 | 0.9432 |
| Tropo | 5th state | >1000 | 1371 | 0.8228 | 0.8016 | 0.9494 | |
| COI | - | 868 | 666 | 0.536 | 0.5118 | 0.8657 |
Acknowledgments
References and Notes
- Futuyma, D; Moreno, G. The Evolution of ecological specialization. Ann. Rev. Ecolog. Syst 1988, 19, 207–233. [Google Scholar]
- Desdevises, Y; Morand, S; Legendre, P. Evolution and determinants of host specificity in the genus Lamellodiscus (Monogenea). Biol. J. Linn. Soc 2002, 77, 431–443. [Google Scholar]
- Danforth, BN; Sipes, S; Fang, J; Brady, SG. The history of early bee diversification based on five genes plus morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 15118–15123. [Google Scholar]
- D’Haese, CA. Were the first springtails semi-aquatic? A phylogenetic approach by means of 28S rDNA and optimization alignment. Proc. Royal Soc.:Biol. Sci 2002, 269, 1143–1151. [Google Scholar]
- Zhong, S; Miller, SP; Dykhuizen, DE; Dean, AM. Transcription, Translation, and the Evolution of Specialists and Generalists. Mol. Biol. Evol 2009, 26, 2661–2678. [Google Scholar]
- Kaci-Chaouch, T; Verneau, O; Desdevises, Y. Host specificity is linked to intraspecific variability in the genus Lamellodiscus (Monogenea). Parasitology 2008, 135, 607–616. [Google Scholar]
- Kuris, AM; Lafferty, KD. Parasite-host modelling meets reality: Adaptive peaks and their ecological attributes. In Evolutionary Biology of Host-Parasite Relationships: Theory Meets Reality; Poulin, R, Skorping, A, Eds.; Elsevier: Amsterdam, The Netherlands, 2000; pp. 9–26. [Google Scholar]
- Moss, WW. The mite genus Dermanyssus: A survey, with description of Dermanyssus trochilinis, n. sp., and a revised key to the species (Acari: Mesostigmata: Dermanyssidae). J. Med. Entomol 1978, 14, 627–640. [Google Scholar]
- Radovsky, FJ. The evolution of parasitism and the distribution of some Dermanyssoid Mites (Mesostigmata) on vertebrate hosts. In Mites Ecological and Evolutionary Analyses of Life-History Patterns; Houck, MA, Ed.; Chapman & Hall: New York, NY, USA, 1994. [Google Scholar]
- Roy, L; Dowling, AP; Chauve, CM; Lesna, I; Sabelis, MW; Buronfosse, T. Molecular phylogenetic assessment of host range in five Dermanyssus species. Exp. Appl. Acarol 2009, 48, 115–142. [Google Scholar]
- Roy, L; Dowling, AP; Chauve, CM; Buronfosse, T. Delimiting species boundaries within Dermanyssus Duges, 1834 (Acari: Dermanyssidae) using a total evidence approach. Mol. Phylogenet. Evol 2009, 50, 446–470. [Google Scholar]
- De Lillo, E. A modified method for Eriophyoid mite extraction (Acari: Eriophyoidea). Internat. J. Acarol 2001, 27, 67–70. [Google Scholar]
- Oliver, JH, Jr. Notes on reproductive behavior in the Dermanyssidae (Acarina Mesostigmata). J. Med. Entomol 1966, 3, 29–35. [Google Scholar]
- Hutcheson, HJ; Oliver, JH, Jr. Spermiogenesis and reproductive biology of Dermanyssus gallinae (DeGeer) (Parasitiformes: Dermanyssidae). J. Med. Entomol 1988, 25, 321–330. [Google Scholar]
- Edgar, R. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004, 32, 1792–1797. [Google Scholar]
- Galtier, N; Gouy, M; Gautier, C. SEAVIEW and PHYLO_WIN: Two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci 1996, 12, 543–548. [Google Scholar]
- Li, B; Lecointre, G. Formalizing reliability in the taxonomic congruence approach. Zool. Scr 2009, 38, 101–112. [Google Scholar]
- Rambaut, A. Drummond, Tracer v1.4. 2009. Available at: http://tree.bio.ed.ac.uk/software/tracer (Accessed on 15 December 2007).
- Birky, CW; Fuerst, P; Maruyama, T. Organelle gene diversity under migration, mutation and drift: Equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to nuclear genes. Genetics 1989, 121, 613–627. [Google Scholar]
- Rozas, J; Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci 1995, 11, 621–625. [Google Scholar]
- Excoffier, L; Laval, G; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinf. Online 2005, 1, 47–50. [Google Scholar]
- Black, WC; Piesman, J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc. Natl. Acad. Sci. USA 1994, 91, 10034–10038. [Google Scholar]
- Sanchis, A; Michelena, JM; Latorre, A; Quicke, DL; Gardenfors, U; Belshaw, R. The phylogenetic analysis of variable-length sequence data: Elongation factor-1alpha introns in European populations of the parasitoid wasp genus Pauesia (Hymenoptera: Braconidae: Aphidiinae). Mol. Biol. Evol 2001, 18, 1117–1131. [Google Scholar]
- Kawakita, A; Sota, T; Ascher, JS; Ito, M; Tanaka, H; Kato, M. Evolution and phylogenetic utility of alignment gaps within intron sequences of three nuclear genes in bumble bees (Bombus). Mol. Biol. Evol 2003, 20, 87–92. [Google Scholar]
- Hedin, MC; Maddison, WP. Phylogenetic utility and evidence for multiple copies of elongation factor-1alpha in the spider genus Habronattus (Araneae: Salticidae). Mol. Biol. Evol 2001, 18, 1512–1521. [Google Scholar]
- Nisbet, AJ; Huntley, JF; Mackellar, A; Sparks, N; McDevitt, R. A house dust mite allergen homologue from poultry red mite Dermanyssus gallinae (De Geer). Parasite Immunol 2006, 28, 401–405. [Google Scholar]
- De Rojas, M; Ubeda, JM; Cutillas, C; Mora, MD; Ariza, C; Guevara, D. Utility of ITS1–5.8s-ITS2 and 16S mitochondrial DNA sequences for species identification and phylogenetic inference within the Rhinonyssus coniventris species complex (Acari: Rhinonyssidae). Parasitol. Res 2007, 100, 1041–1046. [Google Scholar]
- De Rojas, M; Mora, MD; Ubeda, JM; Cutillas, C; Navajas, M; Guevara, DC. Phylogenetic relationships in rhinonyssid mites (Acari: Rhinonyssidae) based on ribosomal DNA sequences: Insights for the discrimination of closely related species. Parasitol. Res 2002, 88, 675–681. [Google Scholar]
- Navajas, M; Lagnel, J; Fauvel, G; De Moraes, G. Sequence variation of ribosomal internal transcribed spacers (ITS) in commercially important Phytoseiidae mites. Exp. Appl. Acarol 1999, 23, 851–859. [Google Scholar]
- Brännström, S; Morrison, DA; Mattsson, JG; Chirico, J. Genetic differences in internal transcribed spacer 1 between Dermanyssus gallinae from wild birds and domestic chickens. Med. Vet. Entomol 2008, 22, 152–155. [Google Scholar]
- Hovemann, B; Richter, S; Walldorf, U; Cziepluch, C. Two genes encode related cytoplasmic elongation factor 1-alpha (EF-1alpha) in Drosophila melanogaster with continuous and stage specific expression. Nucleic Acids Res 1988, 16, 3175–3194. [Google Scholar]
- Danforth, BN; Ji, S. Elongation factor-1 alpha occurs as two copies in bees: Implications for phylogenetic analysis of EF-1 alpha sequences in insects. Mol. Biol. Evol 1998, 15, 225–235. [Google Scholar]
- Williams, ST; Knowlton, N; Weigt, LA; Jara, JA. Evidence for three major clades within the snapping shrimp genus Alpheus inferred from nuclear and mitochondrial gene sequence data. Mol. Phylogenet. Evol 2001, 20, 375–389. [Google Scholar]
- Goetze, E. Elongation factor 1-alpha in marine copepods (Calanoida: Eucalanidae): Phylogenetic utility and unique intron structure. Mol. Phylogenet. Evol 2006, 40, 880–886. [Google Scholar]
- Brady, SG; Danforth, BN. Recent intron gain in elongation factor-1alpha of colletid bees (Hymenoptera: Colletidae). Mol. Biol. Evol 2004, 21, 691–696. [Google Scholar]
- Klompen, H. Preliminary Assessment of the utility of elongation factor-1alpha in elucidating relationships among basal Mesostigmata. Exp. Appl. Acarol 2000, 24, 805–820. [Google Scholar]
- Klompen, H; Lekveishvili, M; Black, WC. Phylogeny of parasitiform mites (Acari) based on rRNA. Mol. Phylogenet. Evol 2007, 43, 936–951. [Google Scholar]
- Nichols, R. Gene trees and species trees are not the same. Trends Ecol. Evol 2000, 16, 358–364. [Google Scholar]
- McCracken, K; Sorenson, M. Is homoplasy or lineage sorting the source of incongruent mtDNA and nuclear gene trees in the stiff-tailed ducks (Nomonyx-oxyura)? Syst. Biol 2005, 54, 35–55. [Google Scholar]
- Moore, WS. Inferring phylogenies from the mt-DNA variation: Mitochondrial-gene trees versus nuclear-gene trees. Evolution 1995, 49, 718–726. [Google Scholar]
- Michaux, JR; Chevret, P; Filippucci, MG; Macholan, M. Phylogeny of the genus Apodemus with a special emphasis on the subgenus Sylvaemus using the nuclear IRBP gene and two mitochondrial markers: Cytochrome b and 12SrRNA. Mol. Phylogenet. Evol 2002, 23, 123–136. [Google Scholar]
- Moss, WW. An Illustrated Key to the Species of the Acarine Genus Dermanyssus (Mesostigmata: Laelapoidea: Dermanyssidae). J. Med. Entomol 1968, 1, 67–84. [Google Scholar]
- Peterson, A. Zoonomen Nomenclatural Data, Version 8.07.
- Seehausen, O; Takimoto, G; Roy, D; Jokela, J. Speciation reversal and biodiversity dynamics with hybridization in changing environments. Mol. Ecol 2007, 17, 30–44. [Google Scholar]
- Whitney, KD; Ahern, JR; Campbell, LG. Hybridization-prone plant families do not generate more invasive species. Biol. Invas 2009, 11, 1205–1215. [Google Scholar]
- Peccoud, J; Ollivier, A; Plantegenest, M; Simon, JC. A continuum of genetic divergence from sympatric host races to species in the pea aphid complex. Proc. Natl. Acad. Sci. USA 2009, 106, 7495–7500. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roy, L.; Dowling, A.P.G.; Chauve, C.M.; Buronfosse, T. Diversity of Phylogenetic Information According to the Locus and the Taxonomic Level: An Example from a Parasitic Mesostigmatid Mite Genus. Int. J. Mol. Sci. 2010, 11, 1704-1734. https://doi.org/10.3390/ijms11041704
Roy L, Dowling APG, Chauve CM, Buronfosse T. Diversity of Phylogenetic Information According to the Locus and the Taxonomic Level: An Example from a Parasitic Mesostigmatid Mite Genus. International Journal of Molecular Sciences. 2010; 11(4):1704-1734. https://doi.org/10.3390/ijms11041704
Chicago/Turabian StyleRoy, Lise, Ashley P. G. Dowling, Claude Marie Chauve, and Thierry Buronfosse. 2010. "Diversity of Phylogenetic Information According to the Locus and the Taxonomic Level: An Example from a Parasitic Mesostigmatid Mite Genus" International Journal of Molecular Sciences 11, no. 4: 1704-1734. https://doi.org/10.3390/ijms11041704
APA StyleRoy, L., Dowling, A. P. G., Chauve, C. M., & Buronfosse, T. (2010). Diversity of Phylogenetic Information According to the Locus and the Taxonomic Level: An Example from a Parasitic Mesostigmatid Mite Genus. International Journal of Molecular Sciences, 11(4), 1704-1734. https://doi.org/10.3390/ijms11041704