Intrinsically Disordered Proteins in Bcl-2 Regulated Apoptosis

Abstract
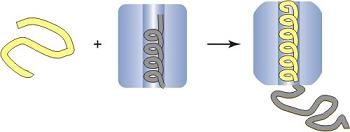
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
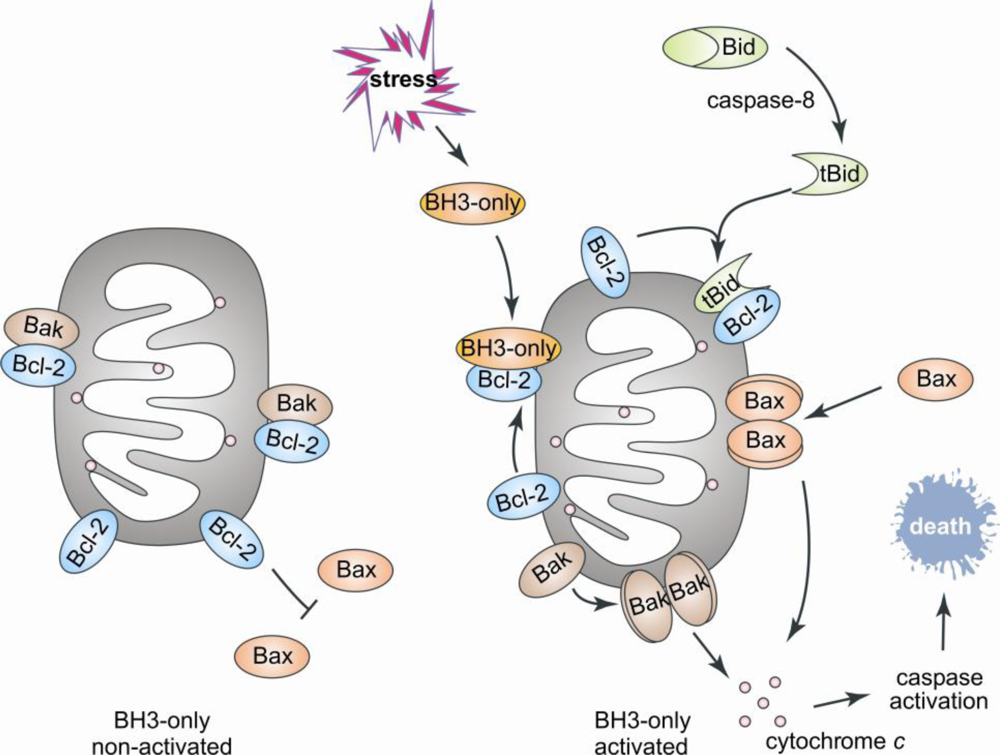
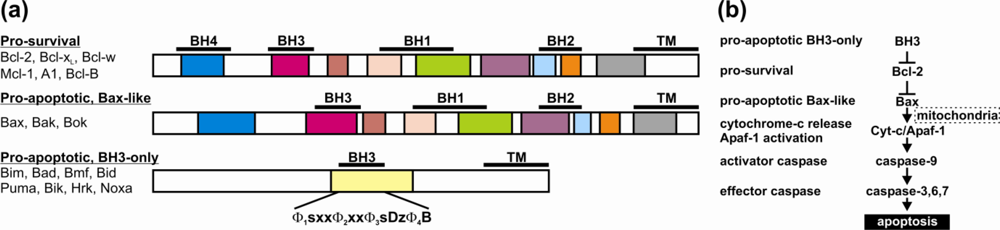
2. Bcl-2 Proteins Are IDPs, or Contain IDRs
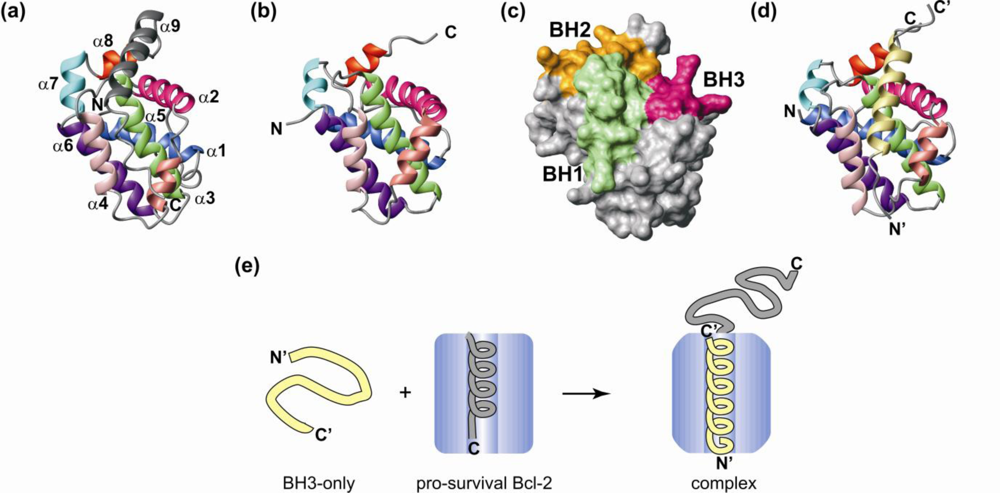
3. Structural Transitions Characterize Bcl-2 Interactions
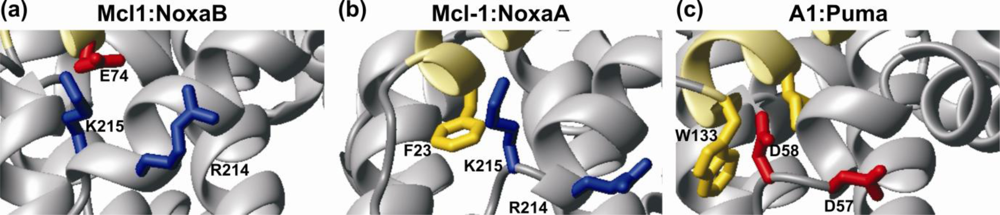
4. Bcl-2 Protein Structural Plasticity and Multiple Binding Partners
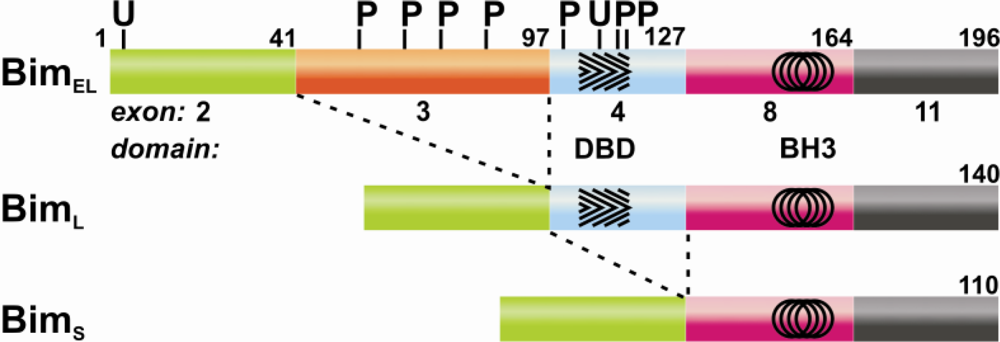
5. Alternate Splicing Leads to Structural Diversity
6. Generating New IDRs and Reactivity through Post-translational Modification
7. Conclusions
Acknowledgments
References and Notes
- Youle, RJ; Strasser, A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell. Biol 2008, 9, 47–59. [Google Scholar]
- Willis, SN; Adams, JM. Life in the balance: How BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol 2005, 17, 617–625. [Google Scholar]
- Hinds, MG; Day, CL. Regulation of apoptosis: Uncovering the binding determinants. Curr. Opin. Struct. Biol 2005, 15, 690–699. [Google Scholar]
- Schinzel, A; Kaufmann, T; Borner, C. Bcl-2 family members: Integrators of survival and death signals in physiology and pathology. Biochim. Biophys. Acta 2004, 1644, 95–105. [Google Scholar]
- Antignani, A; Youle, RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol 2006, 18, 685–689. [Google Scholar]
- Wei, MC; Zong, WX; Cheng, EH; Lindsten, T; Panoutsakopoulou, V; Ross, AJ; Roth, KA; MacGregor, GR; Thompson, CB; Korsmeyer, SJ. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar]
- Hanahan, D; Weinberg, RA. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Fesik, SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 2005, 5, 876–885. [Google Scholar]
- Walensky, LD; Kung, AL; Escher, I; Malia, TJ; Barbuto, S; Wright, RD; Wagner, G; Verdine, GL; Korsmeyer, SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar]
- Muchmore, SW; Sattler, M; Liang, H; Meadows, RP; Harlan, JE; Yoon, HS; Nettesheim, D; Chang, BS; Thompson, CB; Wong, SL; Ng, SL; Fesik, SW. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996, 381, 335–341. [Google Scholar]
- Sattler, M; Liang, H; Nettesheim, D; Meadows, RP; Harlan, JE; Eberstadt, M; Yoon, HS; Shuker, SB; Chang, BS; Minn, AJ; Thompson, CB; Fesik, SW. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar]
- Aritomi, M; Kunishima, N; Inohara, N; Ishibashi, Y; Ohta, S; Morikawa, K. Crystal structure of rat Bcl-xL. Implications for the function of the Bcl-2 protein family. J. Biol. Chem 1997, 272, 27886–27892. [Google Scholar]
- Blaineau, SV; Aouacheria, A. BCL2DB: Moving ‘helix-bundled’ BCL-2 family members to their database. Apoptosis 2009, 14, 923–925. [Google Scholar]
- Suzuki, M; Youle, RJ; Tjandra, N. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell 2000, 103, 645–654. [Google Scholar]
- Denisov, AY; Madiraju, MS; Chen, G; Khadir, A; Beauparlant, P; Attardo, G; Shore, GC; Gehring, K. Solution structure of human BCL-w: Modulation of ligand binding by the C-terminal helix. J. Biol. Chem 2003, 278, 21124–21128. [Google Scholar]
- Hinds, MG; Lackmann, M; Skea, GL; Harrison, PJ; Huang, DC; Day, CL. The structure of Bcl-w reveals a role for the C-terminal residues in modulating biological activity. EMBO J 2003, 22, 1497–1507. [Google Scholar]
- Day, CL; Chen, L; Richardson, SJ; Harrison, PJ; Huang, DC; Hinds, MG. Solution Structure of Prosurvival Mcl-1 and Characterization of Its Binding by Proapoptotic BH3-only Ligands. J. Biol. Chem 2005, 280, 4738–4744. [Google Scholar]
- Germain, M; Duronio, V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. J. Biol. Chem 2007, 282, 32233–32242. [Google Scholar]
- Rogers, S; Wells, R; Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science 1986, 234, 364–368. [Google Scholar]
- De Biasio, A; Vrana, JA; Zhou, P; Qian, L; Bieszczad, CK; Braley, KE; Domina, AM; Weintraub, SJ; Neveu, JM; Lane, WS; Craig, RW. N-terminal truncation of antiapoptotic MCL1, but not G2/M-induced phosphorylation, is associated with stabilization and abundant expression in tumor cells. J. Biol. Chem 2007, 282, 23919–23936. [Google Scholar]
- Hinds, MG; Smits, C; Fredericks-Short, R; Risk, JM; Bailey, M; Huang, DC; Day, CL. Bim, Bad and Bmf: Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ 2007, 14, 128–136. [Google Scholar]
- Liu, X; Dai, S; Zhu, Y; Marrack, P; Kappler, JW. The structure of a Bcl-xL/Bim fragment complex: Implications for Bim function. Immunity 2003, 19, 341–352. [Google Scholar]
- McDonnell, JM; Fushman, D; Milliman, CL; Korsmeyer, SJ; Cowburn, D. Solution structure of the proapoptotic molecule BID: A structural basis for apoptotic agonists and antagonists. Cell 1999, 96, 625–634. [Google Scholar]
- Chou, JJ; Li, H; Salvesen, GS; Yuan, J; Wagner, G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 1999, 96, 615–624. [Google Scholar]
- Yao, Y; Bobkov, AA; Plesniak, LA; Marassi, FM. Mapping the interaction of pro-apoptotic tBID with pro-survival BCL-xL. Biochemistry 2009, 48, 8704–8711. [Google Scholar]
- Smits, C; Czabotar, PE; Hinds, MG; Day, CL. Structural plasticity underpins promiscuous binding of the pro-survival protein A1. Structure 2008, 16, 818–829. [Google Scholar]
- Day, CL; Smits, C; Fan, FC; Lee, EF; Fairlie, WD; Hinds, MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J. Mol. Biol 2008, 380, 958–971. [Google Scholar]
- Lee, EF; Czabotar, PE; Yang, H; Sleebs, BE; Lessene, G; Colman, PM; Smith, BJ; Fairlie, WD. Conformational changes in Bcl-2 pro-survival proteins determine their capacity to bind ligands. J. Biol. Chem 2009, 284, 30508–30517. [Google Scholar]
- Gavathiotis, E; Suzuki, M; Davis, ML; Pitter, K; Bird, GH; Katz, SG; Tu, HC; Kim, H; Cheng, EH; Tjandra, N; Walensky, LD. BAX activation is initiated at a novel interaction site. Nature 2008, 455, 1076–1081. [Google Scholar]
- Hsu, YT; Wolter, KG; Youle, RJ. Cytosol-to-membrane redistribution of Bax and Bcl-xL during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar]
- Wilson-Annan, J; O’Reilly, LA; Crawford, SA; Hausmann, G; Beaumont, JG; Parma, LP; Chen, L; Lackmann, M; Lithgow, T; Hinds, MG; Day, CL; Adams, JM; Huang, DC. Proapoptotic BH3-only proteins trigger membrane integration of prosurvival Bcl-w and neutralize its activity. J. Cell Biol 2003, 162, 877–887. [Google Scholar]
- Certo, M; Del Gaizo Moore, V; Nishino, M; Wei, G; Korsmeyer, S; Armstrong, SA; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar]
- Kuwana, T; Bouchier-Hayes, L; Chipuk, JE; Bonzon, C; Sullivan, BA; Green, DR; Newmeyer, DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar]
- Chen, L; Willis, SN; Wei, A; Smith, BJ; Fletcher, JI; Hinds, MG; Colman, PM; Day, CL; Adams, JM; Huang, DC. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar]
- Leung, DH; Bergman, RG; Raymond, KN. Enthalpy-entropy compensation reveals solvent reorganization as a driving force for supramolecular encapsulation in water. J. Am. Chem. Soc 2008, 130, 2798–2805. [Google Scholar]
- Dyson, HJ; Wright, PE. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol 2002, 12, 54–60. [Google Scholar]
- Wright, PE; Dyson, HJ. Linking folding and binding. Curr. Opin. Struct. Biol 2009, 19, 31–38. [Google Scholar]
- Lee, EF; Czabotar, PE; Smith, BJ; Deshayes, K; Zobel, K; Colman, PM; Fairlie, WD. Crystal structure of ABT-737 complexed with Bcl-xL: Implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ 2007, 14, 1711–1713. [Google Scholar]
- Lee, EF; Fedorova, A; Zobel, K; Boyle, MJ; Yang, H; Perugini, MA; Colman, PM; Huang, DC; Deshayes, K; Fairlie, WD. Novel BCL-2 homology BH3 domain-like sequences identified from screening randomized peptide libraries for inhibitors of the pro-survival BCL 2 proteins. J. Biol. Chem 2009, 284, 31315–31326. [Google Scholar]
- Fire, E; Gulla, SV; Grant, RA; Keating, AE. Mcl-1-Bim complexes accommodate surprising point mutations via minor structural changes. Protein Sci 2010, 19, 507–519. [Google Scholar]
- Akgul, C; Moulding, DA; Edwards, SW. Alternative splicing of Bcl-2-related genes: Functional consequences and potential therapeutic applications. Cell Mol. Life. Sci 2004, 61, 2189–2199. [Google Scholar]
- Huang, DC; Strasser, A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell 2000, 103, 839–842. [Google Scholar]
- Aouacheria, A; Brunet, F; Gouy, M. Phylogenomics of life-or-death switches in multicellular animals: Bcl-2, BH3-Only, and BNip families of apoptotic regulators. Mol. Biol. Evol 2005, 22, 2395–2416. [Google Scholar]
- O’Connor, L; Strasser, A; O’Reilly, LA; Hausmann, G; Adams, JM; Cory, S; Huang, DC. Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J 1998, 17, 384–395. [Google Scholar]
- Bouillet, P; Zhang, LC; Huang, DC; Webb, GC; Bottema, CD; Shore, P; Eyre, HJ; Sutherland, GR; Adams, JM. Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm. Genome 2001, 12, 163–168. [Google Scholar]
- Marani, M; Tenev, T; Hancock, D; Downward, J; Lemoine, NR. Identification of novel isoforms of the BH3 domain protein Bim which directly activate Bax to trigger apoptosis. Mol. Cell Biol 2002, 22, 3577–3589. [Google Scholar]
- Renshaw, SA; Dempsey, CE; Barnes, FA; Bagstaff, SM; Dower, SK; Bingle, CD; Whyte, MK. Three novel Bid proteins generated by alternative splicing of the human Bid gene. J. Biol. Chem 2004, 279, 2846–2855. [Google Scholar]
- Hamner, S; Arumae, U; Li-Ying, Y; Sun, YF; Saarma, M; Lindholm, D. Functional characterization of two splice variants of rat bad and their interaction with Bcl-w in sympathetic neurons. Mol. Cell Neurosci 2001, 17, 97–106. [Google Scholar]
- Seo, SY; Chen, YB; Ivanovska, I; Ranger, AM; Hong, SJ; Dawson, VL; Korsmeyer, SJ; Bellows, DS; Fannjiang, Y; Hardwick, JM. BAD is a pro-survival factor prior to activation of its pro-apoptotic function. J. Biol. Chem 2004, 279, 42240–42249. [Google Scholar]
- Morales, AA; Olsson, A; Celsing, F; Osterborg, A; Jondal, M; Osorio, LM. Expression and transcriptional regulation of functionally distinct Bmf isoforms in B-chronic lymphocytic leukemia cells. Leukemia 2004, 18, 41–47. [Google Scholar]
- Gomez-Esquer, F; Palomar, MA; Rivas, I; Delcan, J; Linares, R; Diaz-Gil, G. Characterization of the BH3 protein Bmf in Gallus gallus: Identification of a novel chicken-specific isoform. Gene 2008, 407, 21–29. [Google Scholar]
- Nakano, K; Vousden, KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar]
- Wang, Z; Sun, Y. Identification and characterization of two splicing variants of human Noxa. Anticancer Res 2008, 28, 1667–1674. [Google Scholar]
- Willis, SN; Chen, L; Dewson, G; Wei, A; Naik, E; Fletcher, JI; Adams, JM; Huang, DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 2005, 19, 1294–1305. [Google Scholar]
- Puthalakath, H; Huang, DC; O’Reilly, LA; King, SM; Strasser, A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell 1999, 3, 287–296. [Google Scholar]
- Puthalakath, H; Villunger, A; O’Reilly, LA; Beaumont, JG; Coultas, L; Cheney, RE; Huang, DC; Strasser, A. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 2001, 293, 1829–1832. [Google Scholar]
- Day, CL; Puthalakath, H; Skea, G; Strasser, A; Barsukov, I; Lian, LY; Huang, DC; Hinds, MG. Localization of dynein light chains 1 and 2 and their pro-apoptotic ligands. Biochem. J 2004, 377, 597–605. [Google Scholar]
- Fan, J; Zhang, Q; Tochio, H; Li, M; Zhang, M. Structural basis of diverse sequence-dependent target recognition by the 8 kDa dynein light chain. J. Mol. Biol 2001, 306, 97–108. [Google Scholar]
- Adachi, M; Zhao, X; Imai, K. Nomenclature of dynein light chain-linked BH3-only protein Bim isoforms. Cell Death Differ 2005, 12, 192–193. [Google Scholar]
- Bae, J; Leo, CP; Hsu, SY; Hsueh, AJ. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J. Biol. Chem 2000, 275, 25255–25261. [Google Scholar]
- Kim, JH; Sim, SH; Ha, HJ; Ko, JJ; Lee, K; Bae, J. MCL-1ES, a novel variant of MCL-1, associates with MCL-1L and induces mitochondrial cell death. FEBS Lett 2009, 583, 2758–2764. [Google Scholar]
- Fu, NY; Sukumaran, SK; Kerk, SY; Yu, VC. Baxbeta: A constitutively active human Bax isoform that is under tight regulatory control by the proteasomal degradation mechanism. Mol. Cell 2009, 33, 15–29. [Google Scholar]
- Romero, PR; Zaidi, S; Fang, YY; Uversky, VN; Radivojac, P; Oldfield, CJ; Cortese, MS; Sickmeier, M; LeGall, T; Obradovic, Z; Dunker, AK. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar]
- Birzele, F; Csaba, G; Zimmer, R. Alternative splicing and protein structure evolution. Nucleic Acids Res 2008, 36, 550–558. [Google Scholar]
- Gsponer, J; Futschik, ME; Teichmann, SA; Babu, MM. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar]
- Puthalakath, H; Strasser, A. Keeping killers on a tight leash: Transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ 2002, 9, 505–512. [Google Scholar]
- Kutuk, O; Letai, A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr. Mol. Med 2008, 8, 102–118. [Google Scholar]
- Danial, NN. BAD: Undertaker by night, candyman by day. Oncogene 2008, 27(Suppl. 1), S53–S70. [Google Scholar]
- Chiang, CW; Kanies, C; Kim, KW; Fang, WB; Parkhurst, C; Xie, M; Henry, T; Yang, E. Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol. Cell Biol 2003, 23, 6350–6362. [Google Scholar]
- Klumpp, S; Selke, D; Krieglstein, J. Protein phosphatase type 2C dephosphorylates BAD. Neurochem. Int 2003, 42, 555–560. [Google Scholar]
- Donovan, N; Becker, EB; Konishi, Y; Bonni, A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem 2002, 277, 40944–40949. [Google Scholar]
- Konishi, Y; Lehtinen, M; Donovan, N; Bonni, A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol. Cell 2002, 9, 1005–1016. [Google Scholar]
- Dramsi, S; Scheid, MP; Maiti, A; Hojabrpour, P; Chen, X; Schubert, K; Goodlett, DR; Aebersold, R; Duronio, V. Identification of a novel phosphorylation site, Ser-170, as a regulator of bad pro-apoptotic activity. J. Biol. Chem 2002, 277, 6399–6405. [Google Scholar]
- Luciano, F; Jacquel, A; Colosetti, P; Herrant, M; Cagnol, S; Pages, G; Auberger, P. Phosphorylation of BimEL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 2003, 22, 6785–6793. [Google Scholar]
- Ley, R; Ewings, KE; Hadfield, K; Howes, E; Balmanno, K; Cook, SJ. Extracellular signal-regulated kinases 1/2 are serum-stimulated “BimEL kinases” that bind to the BH3-only protein BimEL causing its phosphorylation and turnover. J. Biol. Chem 2004, 279, 8837–8847. [Google Scholar]
- Ewings, KE; Hadfield-Moorhouse, K; Wiggins, CM; Wickenden, JA; Balmanno, K; Gilley, R; Degenhardt, K; White, E; Cook, SJ. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J 2007, 26, 2856–2867. [Google Scholar]
- Ley, R; Balmanno, K; Hadfield, K; Weston, C; Cook, SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J. Biol. Chem 2003, 278, 18811–18816. [Google Scholar]
- Qi, XJ; Wildey, GM; Howe, PH. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J. Biol. Chem 2006, 281, 813–823. [Google Scholar]
- Lord, SJ; Rajotte, RV; Korbutt, GS; Bleackley, RC. Granzyme B: A natural born killer. Immunol. Rev 2003, 193, 31–38. [Google Scholar]
- Zha, J; Weiler, S; Oh, KJ; Wei, MC; Korsmeyer, SJ. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 2000, 290, 1761–1765. [Google Scholar]
- Garcia-Saez, AJ; Ries, J; Orzaez, M; Perez-Paya, E; Schwille, P. Membrane promotes tBID interaction with BCL-xL. Nat. Struct. Mol. Biol 2009, 16, 1178–1185. [Google Scholar]
- Desagher, S; Osen-Sand, A; Montessuit, S; Magnenat, E; Vilbois, F; Hochmann, A; Journot, L; Antonsson, B; Martinou, JC. Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol. Cell 2001, 8, 601–611. [Google Scholar]
- Chang, BS; Minn, AJ; Muchmore, SW; Fesik, SW; Thompson, CB. Identification of a novel regulatory domain in Bcl-xL and Bcl-2. EMBO J 1997, 16, 968–977. [Google Scholar]
- Haldar, S; Jena, N; Croce, CM. Inactivation of Bcl-2 by phosphorylation. Proc. Natl. Acad. Sci. USA 1995, 92, 4507–4511. [Google Scholar]
- Yamamoto, K; Ichijo, H; Korsmeyer, SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell Biol 1999, 19, 8469–8478. [Google Scholar]
- Deverman, BE; Cook, BL; Manson, SR; Niederhoff, RA; Langer, EM; Rosova, I; Kulans, LA; Fu, X; Weinberg, JS; Heinecke, JW; Roth, KA; Weintraub, SJ. Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell 2002, 111, 51–62. [Google Scholar]
- Cheng, EH; Kirsch, DG; Clem, RJ; Ravi, R; Kastan, MB; Bedi, A; Ueno, K; Hardwick, JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997, 278, 1966–1968. [Google Scholar]
- Grandgirard, D; Studer, E; Monney, L; Belser, T; Fellay, I; Borner, C; Michel, MR. Alphaviruses induce apoptosis in Bcl-2-overexpressing cells: Evidence for a caspase-mediated, proteolytic inactivation of Bcl-2. EMBO J 1998, 17, 1268–1278. [Google Scholar]
- Yang-Yen, HF. Mcl-1: A highly regulated cell death and survival controller. J. Biomed. Sci 2006, 13, 201–204. [Google Scholar]
- Moulding, DA; Akgul, C; Derouet, M; White, MR; Edwards, SW. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J. Leukoc. Biol 2001, 70, 783–792. [Google Scholar]
- Blagosklonny, MV; Alvarez, M; Fojo, A; Neckers, LM. bcl-2 protein downregulation is not required for differentiation of multidrug resistant HL60 leukemia cells. Leuk. Res 1996, 20, 101–107. [Google Scholar]
- Kozopas, KM; Yang, T; Buchan, HL; Zhou, P; Craig, RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 1993, 90, 3516–3520. [Google Scholar]
- Zhong, Q; Gao, W; Du, F; Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar]
- Deng, X; Kornblau, SM; Ruvolo, PP; May, WS, Jr. Regulation of Bcl-2 phosphorylation and potential significance for leukemic cell chemoresistance. J. Natl. Cancer Inst. Monogr 2001, 28, 30–37. [Google Scholar]
- Meszaros, B; Tompa, P; Simon, I; Dosztanyi, Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol 2007, 372, 549–561. [Google Scholar]





© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rautureau, G.J.P.; Day, C.L.; Hinds, M.G. Intrinsically Disordered Proteins in Bcl-2 Regulated Apoptosis. Int. J. Mol. Sci. 2010, 11, 1808-1824. https://doi.org/10.3390/ijms11041808
Rautureau GJP, Day CL, Hinds MG. Intrinsically Disordered Proteins in Bcl-2 Regulated Apoptosis. International Journal of Molecular Sciences. 2010; 11(4):1808-1824. https://doi.org/10.3390/ijms11041808
Chicago/Turabian StyleRautureau, Gilles J. P., Catherine L. Day, and Mark G. Hinds. 2010. "Intrinsically Disordered Proteins in Bcl-2 Regulated Apoptosis" International Journal of Molecular Sciences 11, no. 4: 1808-1824. https://doi.org/10.3390/ijms11041808
APA StyleRautureau, G. J. P., Day, C. L., & Hinds, M. G. (2010). Intrinsically Disordered Proteins in Bcl-2 Regulated Apoptosis. International Journal of Molecular Sciences, 11(4), 1808-1824. https://doi.org/10.3390/ijms11041808