An Overview of Recent Development in Composite Catalysts from Porous Materials for Various Reactions and Processes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- ♦ Dual-function effect in elementary reactions [43].
2. Amorphous Oxide Modified Zeolite Composite Catalysts
2.1. Amorphous Silica Decorated Zeolite Composite Catalysts
2.2. Phosphorus Oxide Modified Zeolite Composite Catalysts
2.3. Metal Oxide Decorated Zeolite Composite Catalysts
3. Composite Zeolites by Co-Crystallization or Overgrowth
4. Hierarchical Porous Composite Catalysts
5. Host-Guest Porous Composite Catalysts
6. Inorganic and Organic Mesoporous Composite Catalysts with Sulfonic Acid Functionality
7. Polymer/CNT Composite Catalysts
8. Conclusions and Outlook
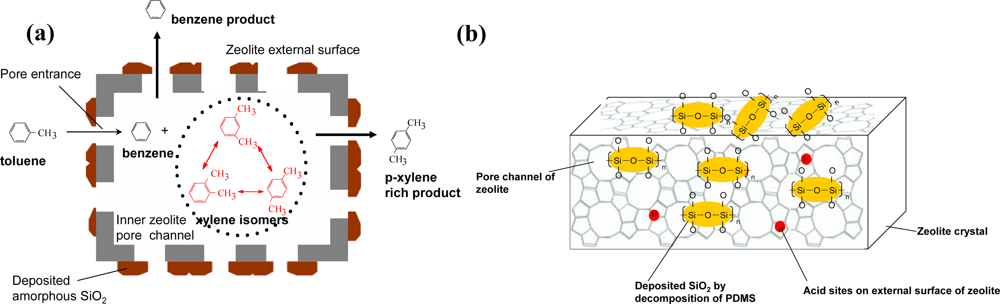
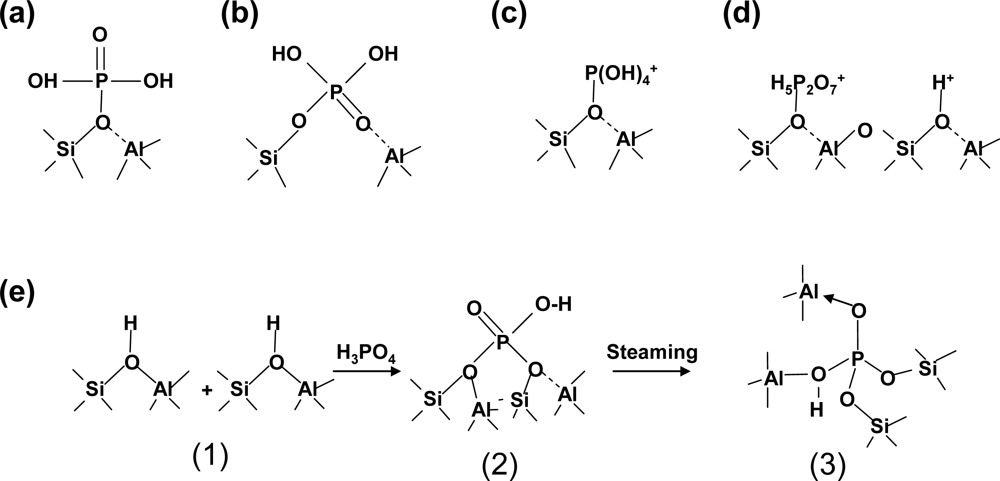
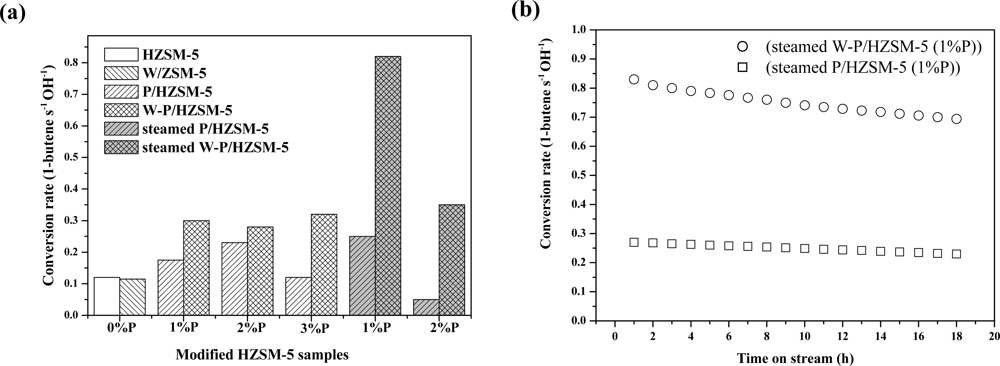
- ✧ Regarding amorphous oxide modified zeolite composite catalysts, we have studied amorphous silica decorated zeolite composite catalysts, phosphorus oxide modified zeolite composite catalysts and metal oxide modified zeolite composite catalysts, among which the former are used for selective toluene disproportionation, and the latter two are used for C4-olefin cracking to produce propylene. The amorphous silica deposition by SiO2-CLD method is to eliminate the external acidic sites of zeolite and reduce the entrance size of zeolite, which are crucial to the high selectivity of para-xylene. Because the phosphorus oxide species stabilized by nonframework Al result in a new kind of acid site that is hydrothermally stable, it is a good method for enhancing the anti-coking ability and hydrothermal stability of zeolite composite catalysts. In addition, synergistic catalytic effects have been found by introducing tungsten into phosphorus-modified HZSM-5.
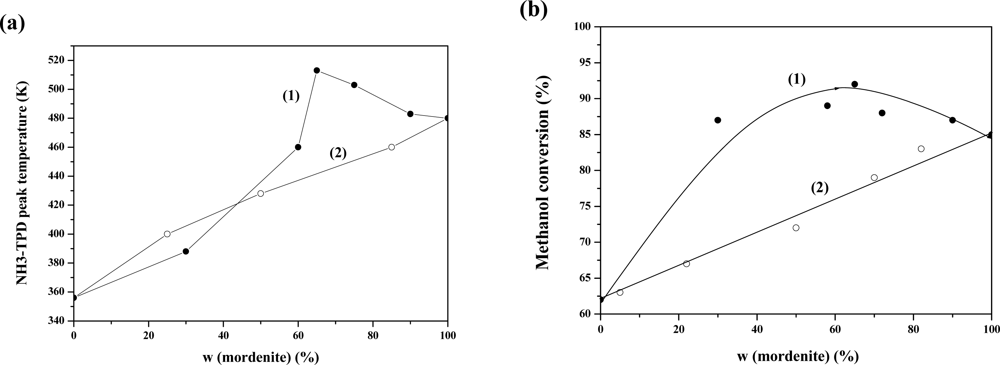
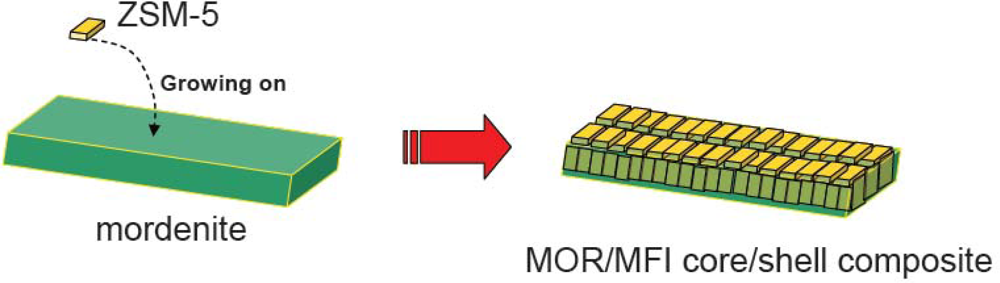
- ✧ Composite zeolites, such as MWW/FER, BEA/MOR and MOR/MFI were synthesized by co-crystallization or overgrowth. It is found that these composite zeolites are somewhat different from mechanical mixtures of the individual zeolites. Characterizations demonstrate that the composite zeolites may be stacked much closer than in the mechanical mixtures, and intergrowth areas may be formed. Compared with mechanical mixtures of zeolites, composite zeolites have higher acid strength and higher activity for acid reactions (aromatization of olefins and methanol dehydration), which may be due to their unique composite structures.
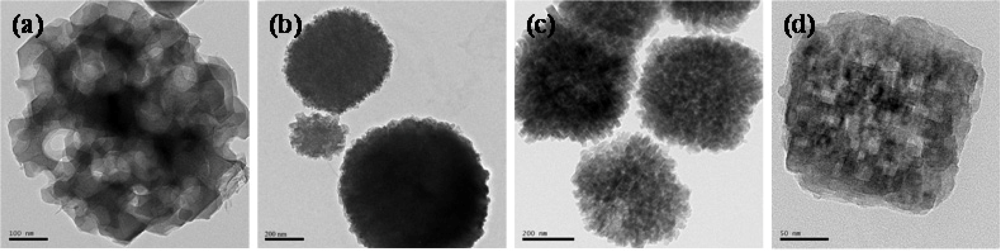
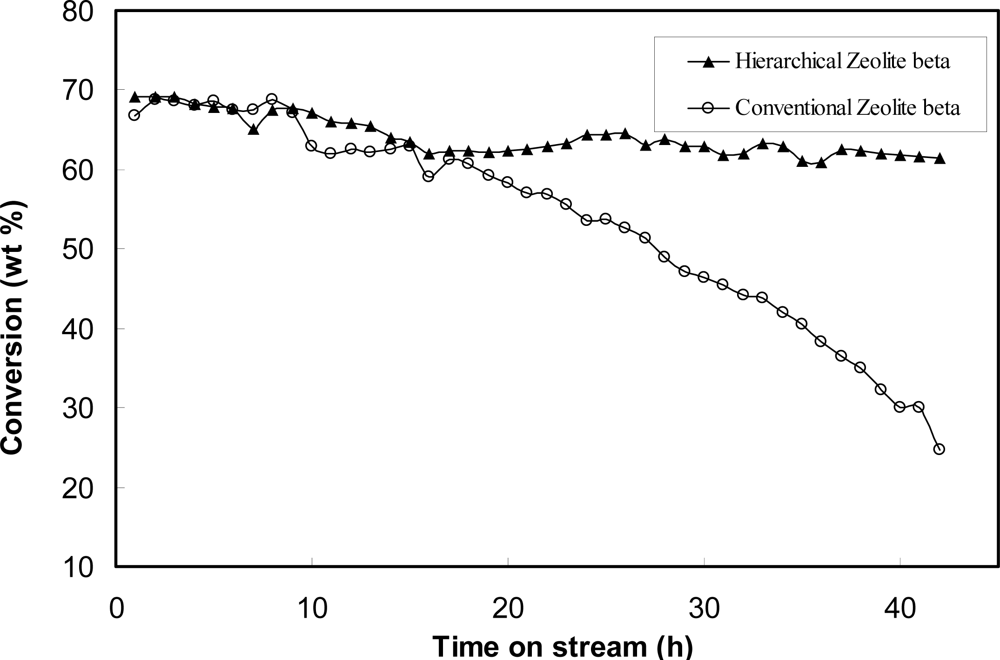
- ✧ As far as hierarchical porous catalysts are concerned, we have tried combine micropores, mesopores and macropores in one catalytic material. Hierarchical porous zeolites, such as silicalite-1, ZSM-5, ZSM-11, Beta were synthesized by using nanosized CaCO3, starch and polyvinyl butyral gel as templates for the generation of mesopores and macropores. It was found that these hierarchical pores were connective with each other which could facilitate the diffusion of molecules. When studying the process of C9 aromatics cracking, more C10 bulky molecules were favorably generated on hierarchical porous zeolites than inside conventional zeolites. Besides, the hierarchical porous zeolites show much lower deactivation rate than conventional zeolites, which could be attributed to the easy transport of molecules with large dimensions through mesopores and macropores.
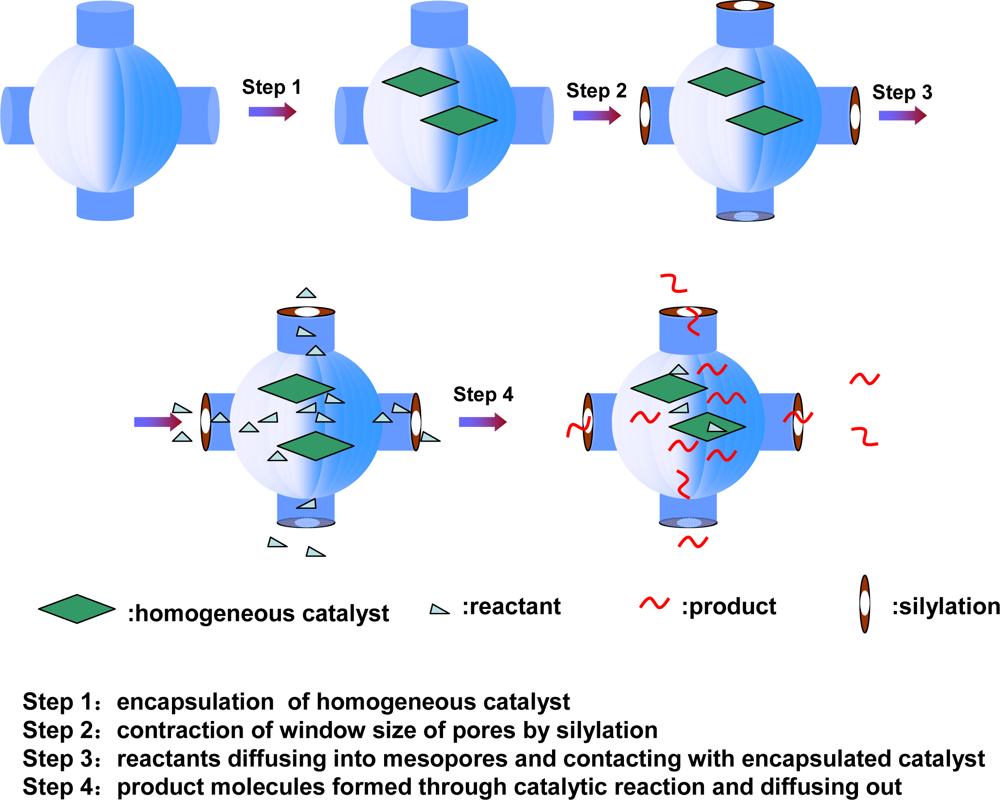
- ✧ As for host-guest porous composites, chiral metal complex catalysts can be trapped in the cage of mesoporous materials like SBA-16 by modifying the entrance pore size of the cage using silylation. The entrapped chiral catalyst can be easily recycled without significant loss of catalytic performance and shows catalytic performance comparable to that in a homogeneous catalysis process. The increase in the activity and enantioselectivity with an increase in the number of [Co(salen)] complexes per cage obviously indicates that the cooperative activation effect of [Co(salen)] complexes in the nanocages can be strengthened owing to the crowded situation of the cobalt complexes in the nanocages.
- ✧ In the field of inorganic and organic mesoporous composite catalysts, we have studied meso-porous organosilica composites with sulfonic acid functionalities. Two methods were applied in preparing such composites: one is a direct synthesis method in which a thiol (-SH) group was oxidized in situ to a sulfonic acid (-SO3H) by hydrogen peroxide; the other is a two-step route in which periodic mesoporous organosilicas containing different fractions of 1,4-diethylene-benzene groups in the framework were synthesized first, and then the sulfonic acid sites were generated by framework sulfonation of 1,4-diethylenebenzene with chlorosulfonic acid. It has been found that these sulfonic acid-functionalized mesoporous organosilicas show high catalytic activity and stability in the liquid-phase condensation of phenol with acetone to form bisphenol A and esterification of ethanol with acetic or other aliphatic acids.
- ✧ As swelling of polymeric ion exchange resins has been an obstacle to their application as catalysts in the catalytic hydration of ethylene oxide, we prepared resin/CNT composite catalysts to overcome this problem. The addition of CNT into a resin can greatly enhance its thermostability and anti-swelling capability during the reaction of hydration of ethylene oxide. The remarkable stability observed should be ascribed to the incorporation of MWNTs in the cross-linking network and the ability to keep the active site loss at low level.
Acknowledgments
References
- Zhao, YQ; Chen, JX; Zhang, JY. Effects of ZrO2 on the performance of CuO-ZnO-Al2O3/HZSM-5 catalyst for dimethyl ether synthesis from CO2 hydrogenation. J. Nat. Gas Chem 2007, 16, 389–392. [Google Scholar]
- Kim, YH; Irie, H; Hashimoto, K. A visible light-sensitive tungsten carbide/tungsten trioxde composite photocatalyst. Appl Phys Lett 2008, 92, 182107-1–182107-3. [Google Scholar]
- Sevcik, P; Cik, G; Vlna, T; Mackulak, T. Preparation and Properties of a New Composite Photocatalyst Based on Nanosized Titanium Dioxide. In Proceedings of 35th International Conference of the Slovak-Society-of-Chemical-Engineering; Tatranske Matliare: Slovakia; 26-30.05.2008; pp. 249–254. [Google Scholar]
- Feng, DM; Wang, YY; Wang, DZ; Wang, JF. Steam reforming of dimethyl ether over CuO-ZnO-Al2O3-ZrO2 + ZSM-5: A kinetic study. Chem. Eng. J 2009, 146, 477–485. [Google Scholar]
- Liu, HH; Zhao, HJ; Gao, XH; Ma, JT. A novel FCC catalyst synthesized via in situ overgrowth of NaY zeolite on kaolin microspheres for maximizing propylene yield. Catal. Today 2007, 125, 163–168. [Google Scholar]
- Qiu, YJ; Chen, JX; Zhang, JY. Effects of CeO2 and CaO composite promoters on the properties of eggshell Ni/MgO-Al2O3 catalysts for partial oxidation of methane to syngas. React. Kinet. Catal. Lett 2008, 94, 351–357. [Google Scholar]
- Liu, BJ; Zeng, XJ; He, LL; Zhao, Z. Catalytic performance of ZSM-5/MOR composite zeolite catalyst for conversion of mixed C-4 hydrocarbons. Chin. J. Catal 2008, 29, 940–944. [Google Scholar]
- Vu, DV; Miyamoto, M; Nishiyama, N; Egashira, Y; Ueyama, K. Morphology control of Silicalite/HZSM-5 composite catalysts for the formation of para-Xylene. Catal. Lett 2009, 127, 233–238. [Google Scholar]
- Wang, YY; Jin, GQ; Guo, XY. Growth of ZSM-5 coating on biomorphic porous silicon carbide derived from durra. Micro. Meso. Mater 2009, 118, 302–306. [Google Scholar]
- Liang, Y; Li, J; Xu, QC; Hu, RZ; Lin, JD; Liao, DW. Characterization of composite carbon supported PtRu catalyst and its catalytic performance for methanol oxidation. J. Alloys. Comp 2008, 465, 296–304. [Google Scholar]
- Imagawa, H; Tanaka, T; Takahashi, N; Matsunaga, S; Suda, A; Shinjoh, H. Synthesis and characterization of Al2O3 and ZrO2-TiO2 nano-composite as a support for NO storage-reduction catalyst. J. Catal 2007, 251, 315–320. [Google Scholar]
- Kong, LR; Lu, XF; Jin, E; Jiang, S; Wang, C; Zhang, WJ. Templated synthesis of polyaniline nanotubes with Pd nanoparticles attached onto their inner walls and its catalytic activity on the reduction of p-nitroanilinum. Compos. Sci. Technol 2009, 69, 561–566. [Google Scholar]
- Long, Q; Cai, M; Rogers, JD; Rong, HL; Li, JR; Jiang, L. Synthesis of nanostructured Pt/CeO2-ZrO2-Al2O3 catalysts by a two-step sol-gel method. Nanotechnology 2007, 18, 355601–355606. [Google Scholar]
- Deng, S; Li, HQ; Li, SG; Zhang, Y. Activity and characterization of modified Cr2O3/ZrO2 nano-composite catalysts for oxidative dehydrogenation of ethane to ethylene with CO2. J. Mol. Catal. A-Chem 2007, 268, 169–175. [Google Scholar]
- Zhao, YQ; Chen, JX; Zhang, JY. Effects of ZrO2 on the performance of CuO-ZnO-Al2O3/HZSM-5 catalyst for dimethyl ether synthesis from CO2 hydrogenation. J. Nat. Gas Chem 2007, 16, 389–392. [Google Scholar]
- Sun, H; Wang, H; Zhang, J. Preparation and characterization of nickel-titanium composite xerogel catalyst for CO2 reforming of CH4. Appl. Catal. B-Environ 2007, 73, 158–165. [Google Scholar]
- Abecassis-Wolfovich, M; Landau, MV; Brenner, A; Herskowitz, M. Low-temperature combustion of 2,4,6-trichlorophenol in catalytic wet oxidation with nanocasted Mn-Ce-oxide catalyst. J. Catal 2007, 247, 201–213. [Google Scholar]
- Ng, YH; Ikeda, S; Harada, T; Park, S; Sakata, T; Mori, H; Matsumura, M. Photocatalytic route for synthesis of hollow porous carbon/Pt nanocomposites with controllable density and porosity. Chem. Mater 2008, 20, 1154–1160. [Google Scholar]
- Nakajima, K; Okamura, M; Kondo, JN; Domen, K; Tatsumi, T; Hayashi, S; Hara, M. Amorphous Carbon Bearing Sulfonic Acid Groups in Mesoporous Silica as a Selective Catalyst. Chem. Mater 2009, 21, 186–193. [Google Scholar]
- Li, L; Shi, JL. Synthesis of rhodium colloidal nano-coating grafted mesoporous silica composite and its application as efficient environmentally benign catalyst for heck-type reaction of arylboronic acids. Adv. Syn. Catal 2008, 350, 667–672. [Google Scholar]
- Zhang, X; Lu, J; Jin, L; Wei, M. Preparation of Rh-TPPTS complex intercalated layered double hydroxide and influences of host and guest compositions on its catalytic performances in hydroformylation reaction. Chin. Sci. Bull 2008, 53, 1329–1336. [Google Scholar]
- Ren, N; Yang, YH; Zhang, YH; Wang, QR; Tang, Y. Heck coupling in zeolitic microcapsular reactor: A test for encaged quasi-homogeneous catalysis. J. Catal 2007, 246, 215–222. [Google Scholar]
- Giri, S; Trewyn, BG; Stellmaker, MP; Lin, VS-Y. Stimuli-responsive controlled-release delivery system based on mesoporous silica nanorods capped with magnetic nanoparticles. Angew. Chem. Int. Ed 2005, 44, 5038–5044. [Google Scholar]
- Lai, C-Y; Trewyn, BG; Jeftinija, DM; Jeftinija, K; Xu, S; Jeftinija, S; Lin, VS-Y. A Mesoporous Silica Nanosphere-Based Carrier System with Chemically Removable CdS Nanoparticle Caps for Stimuli-Responsive Controlled Release of Neurotransmittersand Drug Molecules. J. Am. Chem. Soc 2003, 125, 4451–4459. [Google Scholar]
- Valentini, L; Puglia, D; Carniato, F; Boccaleri, E; Marchese, L; Kenny, JM. Use of plasma fluorinated single-walled carbon nanotubes for the preparation of nanocomposites with epoxy matrix. Compos. Sci. Technol 2008, 68, 1008–1014. [Google Scholar]
- Fa, WJ; Zan, L; Gong, CQ; Zhong, JC; Deng, KJ. Solid-phase photocatalytic degradation of polystyrene with TiO2 modified by iron(II) phthalocyanine. Appl. Catal. B-Environ 2008, 79, 216–223. [Google Scholar]
- Selvaraj, V; Alagar, M; Kumar, KS. Synthesis and characterization of metal nanoparticles-decorated PPY-CNT composite and their electrocatalytic oxidation of formic acid and formaldehyde for fuel cell applications. Appl. Catal. B-Environ 2007, 75, 129–138. [Google Scholar]
- Tang, QW; Lin, JM; Wu, ZB; Wu, JH; Huang, ML; Yang, YY. Preparation and photocatalytic degradability of TiO2/polyacrylamide composite. Eur. Poly. J 2007, 43, 2214–2220. [Google Scholar]
- Perez-Ramirez, J; Christensen, CH; Egeblad, K; Christensend, CH; Groenef, JC. Hierarchical zeolites: Enhanced utilisation of microporous crystals in catalysis by advances in materials design. Chem. Soc. Rev 2008, 37, 2530–2542. [Google Scholar]
- van Donk, S; Janssen, AH; Bitter, JH; de Jong, KP. Generation, characterization, and impact of mesopores in zeolite catalysts. Chem. Rev 2003, 45, 297–319. [Google Scholar]
- Yousheng, T; Hirofumi, K; Lloyd, A; Katsumi, K. Mesopore-modified zeolites: Preparation, characterization, and applications. Catal. Rev 2006, 106, 896–910. [Google Scholar]
- Stambuli, JP; Stauffer, SR; Shaughnessy, KH; Hartwig, JF. Screening of homogeneous catalysts by fluorescence resonance energy transfer. Identification of catalysts for room-temperature heck reactions. J. Am. Chem. Soc 2001, 123, 2677–2678. [Google Scholar]
- Fabing, S; Lu, L; Fang, YL; Tao, L; Andrew, IC; Xiu, SZ. Thermally reduced ruthenium nanoparticles as a highly active heterogeneous catalyst for hydrogenation of monoaromatics. J. Am. Chem. Soc 2007, 45, 11–20. [Google Scholar]
- Escard, J; Leclere, C; Contour, JP. The state of supported irdium in a hydrazine decomposition catalyst. J. Catal 1973, 29, 31–36. [Google Scholar]
- Delgass, WN; Hughes, R; Fadley, CS. X-ray photoelectron spectroscopy: A tool for research in catalysis. Catal. Rev.Sci. Eng 1971, 4, 179–220. [Google Scholar]
- Giordano, N; Bart, JCT; Vaghi, A; Castellan, A; Martinotti, G. Structure and catalytic activity of MoO3 · Al2O3 systems: I. Solid-state properties of oxidized catalysts. J. Catal 1975, 36, 81–92. [Google Scholar]
- Yoshida, S; Iguchi, T; Ishida, S; Tarama, K. Some physico-chemical properties of vanadium oxide supported on silica or γ-alumina. Bull. Chem. Soc. Jpn 1972, 45, 376–380. [Google Scholar]
- Trimm, DL; Doerr, LA. The catalytic oxidative dimerization of propylene. J. Catal 1971, 23, 49–53. [Google Scholar]
- Seiyama, T; Egashira, M; Sakamoto, T; Aso, I. Oxidative dehydroaromatization: II. Oxidation of propylene over binary oxide catalysts containing bismuth or tin. J. Catal 1972, 24, 76–81. [Google Scholar]
- Khalafalla, SE; Haas, LA. The role of metallic component in the iron-alumina bifunctional catalyst for reduction of SO2 with CO. J. Catal 1972, 24, 121–129. [Google Scholar]
- Bond, GC. Heterogeneous Catalysis; Clarendon Press: Oxford, UK, 1974; p. 85. [Google Scholar]
- Mills, GA; Heinemann, H; Milliken, TH; Oblad, AG. (Houdriforming Reactions) Catalytic mechanism. Ind. Eng. Chem 1953, 45, 134–137. [Google Scholar]
- Niiyama, H; Echigoya, E. Hydrogen transfer reaction between alcohols and acetone. Bull.Chem. Soc. Jpn 1972, 45, 938–939. [Google Scholar]
- Benesi, HA. Acidity of Catalyst Surfaces. II. Amine Titration Using Hammett Indicators. J. Phys. Chem 1957, 61, 970–973. [Google Scholar]
- Bridges, JM; Rymer, GT; Maclver, DS. The mechanism of potassium promotion of chromia-alumina dehydrogenation catalysts. J. Phys. Chem 1962, 66, 871–877. [Google Scholar]
- Thomos, CL. Chemistry of cracking catalysts. Ind. Eng. Chem 1949, 41, 2564–2573. [Google Scholar]
- Okamoto, Y; Imanaka, T; Teranishi, S. The Isomerization of Propylene Oxide on Metal Oxides and Silica-Magnesia Catalysts. Bull. Chem. Soc. Jpn 1973, 46, 4–8. [Google Scholar]
- Margolis, LYa. Catalytic oxidation of hydrocarbons. Adv. Catal 1963, 14, 429–501. [Google Scholar]
- Lipshe, JMJG; Schuit, GCA. The CoO-MoO3-Al2O3 catalyst: III. Catalytic properties. J. Catal 1969, 15, 179–189. [Google Scholar]
- Yao, YY. The oxidation of hydrocarbons and CO over metal oxides: IV. Perovskite-type oxides. J. Catal 1975, 36, 266–275. [Google Scholar]
- Aykan, K; Halvorson, D; Sleight, AW; Rogers, DB. Olefin oxidation and ammoxidation studies over molybdate, tungstate, and vanadate catalysts having point defects. J. Catal 1975, 35, 401–406. [Google Scholar]
- Evnin, AB; Rabo, JA; Kasai, PH. Heterogeneously catalyzed vapor-phase oxidation of ethylene to acetaldehyde. J. Catal 1973, 30, 109–117. [Google Scholar]
- Biloen, P; Pott, GT. X-ray photoelectron spectroscopy study of supported tungsten oxide. J. Catal 1973, 30, 169–174. [Google Scholar]
- Adkins, H; Burgoyne, EE; Schneider, HG. The Copper-Chromium Oxide Catalyst for Hydrogenation1. J. Am. Chem. Soc 1950, 72, 2626–2629. [Google Scholar]
- Takita, Y; Ozaki, A; Moro-oka, Y. Catalytic oxidation of olefins over oxide catalysts containing molybdenum: V. Relation between the surface concentration of acidic sites and the catalytic activity to form acetone. J. Catal 1972, 27, 185–192. [Google Scholar]
- Kortunov, P; Vasenkov, S; Karger, J; Valiullin, R; Gottschalk, P; FeElia, M; Perez, M; Stocker, M; Drescher, B; McKeever, LD; Berger, C; Glaser, R; Weitkamp, J. The Role of Mesopores in Intracrystalline Transport in USY Zeolite: PFG NMR Diffusion Study on Various Length Scales. J. Am. Chem. Soc 2005, 127, 13055–13059. [Google Scholar]
- Groen, JC; Zhu, WD; Brouwer, S; Huynink, SJ; Kapteijn, F; Moulijn, JA; Perez-Ramirez, J. Direct Demonstration of Enhanced Diffusion in Mesoporous ZSM-5 Zeolite Obtained via Controlled Desilication. J. Am. Chem. Soc 2007, 129, 355–360. [Google Scholar]
- Lei, Q; Zhao, TB; Li, FY; Zhang, LL; Wang, Y. Catalytic cracking of large molecules over hierarchical zeolites. Chem. Commun 2006, 16, 1769–1771. [Google Scholar]
- Claus, JHJ; Schmidt, I; Krogh, A; Wienberg, K; Carlsson, A; Brorsona, M. Catalytic epoxidation of alkenes with hydrogen peroxide over first mesoporous titanium-containing zeolite. Chem. Commun 2000, 21, 2157–2158. [Google Scholar]
- Ruckenstein, E; Krishnan, R; Rai, KN. Oxygen depletion of oxide catalysts. J. Catal 1976, 45, 270–273. [Google Scholar]
- Boreskov, GK. The catalysis of isotopic exchange in molecular oxygen. Adv. Catal 1964, 15, 285–339. [Google Scholar]
- Benson, JE; Kohn, HW; Boudart, M. On the reduction of tungsten trioxide accelerated by platinum and water. J. Catal 1966, 5, 307–313. [Google Scholar]
- Bond, GC; Molloy, LR; Fuller, MJ. Oxidation of carbon monoxide over palladium–tin (IV) oxide catalysts: An example of spillover catalysis. Chem. Commun 1975, 19, 796–797. [Google Scholar]
- Zheng, S; Heydenrych, HR; Jentys, A; Lercher, JA. Influence of Surface Modification on the Acid Site Distribution of HZSM-5. J. Phys. Chem 2002, 106, 9952–9558. [Google Scholar]
- Faramaway, S. Selective toluene-methanol alkylation over modified ZSM-5 zeolite catalysts. Pet. Sci. Technol 1999, 17, 249–271. [Google Scholar]
- Cejka, J; Zilkov, N; Widhterlova, B. Decisive role of transport rate of products for zeolite para-selectivity: Effect of coke deposition and external surface silylation on activity and selectivity of HZSM-5 in alkylation of toluene. Zeolites 1996, 17, 265–271. [Google Scholar]
- Aboul-Gheit, AK; Abdel-Hamid, SM; Emam, EA. Catalytic para-xylene maximization II–alkylation of toluene with methanol on platinum loaded H-ZSM-5 zeolite catalysts prepared via different routes. Appl. Catal. A 1999, 179, 107–115. [Google Scholar]
- Faramawy, S; El-Sabagh, SM; Al-Mehbad, NY. Selective alkylation of toluene with methanol overp-modified H-ZSM-5: Effect of treatment with chromium and nickel. React. Kinet. Catal. Lett 1999, 66, 257–263. [Google Scholar]
- Hibino, T; Niwa, M; Murakami, Y. Modification of HZSM-5 by CVD of Various Silicon Compounds and Generation of Para-Selectivity. J. Catal 1996, 161, 387–392. [Google Scholar]
- Unneberg, E; Kolboe, S. H-[B]-ZSM-5 as catalyst for methanol reactions. Appl. Catal. A 1995, 124, 345–354. [Google Scholar]
- Duearme, V; Vedrine, JC. ZSM-5 and ZSM-11 zeolites: Influence of morphological and chemical parameters on catalytic selectivity and deactivation. Appl. Catal. A 1985, 17, 175–181. [Google Scholar]
- Kaeding, WW; Young, LB; Chu, CC. Shape-selective reactions with zeolite catalysts: IV. Alkylation of toluene with ethylene to produce p-ethyltoluene. J. Catal 1984, 89, 267–273. [Google Scholar]
- Rotman, D. Catalysts-criterion, laroche in alumina pact. Chem. Week 1995, 157, 18. [Google Scholar]
- Chang, CD; Rodewald, PG. Regioselective methylation of toluene to para-xylene. US Patent 5,607,888 1997. [Google Scholar]
- Beck, JS; Olson, DH; McCullen, SB. Selective toluene disproportionation process (STDP) with ex situ selectivated zeolite catalyst. US Patent 5,367,099 1994. [Google Scholar]
- Beck, JS; Kinn, TF; McCullen, SB. High conversion touluene disproportionation with ex situ selectivated zeolite catalysts. US Patent 5,659,098 1997. [Google Scholar]
- Zhu, ZR; Xie, ZK; Chen, QL; Kong, DJ; Li, W; Yang, WM; Li, C. Chemical liquid deposition with polysiloxane of ZSM-5 and its effect on acidity and catalytic properties. Micropor. Mesopor. Mater 2007, 101, 169–175. [Google Scholar]
- Zhang, XJ; Meng, FH. News: The industrial test of SD-01 catalyst of Shanghai Research Institute of Petrochemical Technology has successfully passed. Petroleum Proc. & Petrochemicals 2006, 37, 60. [Google Scholar]
- Yu, SB. Industrial application of catalyst SD-01 for shape-selective disproportionation of toluene. Tianjin Chem. Ind 2007, 20, 34–36. [Google Scholar]
- Dath, JP; Vermeiren, W. Production of olefins. Eur Patent 1,061,116 2000. [Google Scholar]
- Dath, JP; Delorme, L; Grootjans, JF; Vanhaeren, X; Vermeiren, W. Production of propylene. World Patent 9,929,805 1999. [Google Scholar]
- Zhu, XX; Liu, SL; Song, YQ; Xu, LY. Catalytic cracking of C4 alkenes to propene and ethene: Influences of zeolites pore structures and Si/Al2 ratios. Appl. Catal. A 2005, 288, 134–142. [Google Scholar]
- Teng, JW; Zhao, GL; Xie, ZK; Chen, QL. Effect of ZSM-5 Zeolite Crystal Size on Propylene Production from Catalytic Cracking of C1∼ 4. Chin. J. Catal 2004, 25, 602–606. [Google Scholar]
- Bortnovsky, O; Sazama, P; Wichterlova, B. Cracking of pentenes to C2–C4 light olefins over zeolites and zeotypes: Role of topology and acid site strength and concentration. Appl. Catal. A 2005, 287, 203–213. [Google Scholar]
- Zhao, GL; Teng, JW; Zhang, YH; Xie, ZK; Yue, YH; Chen, QL; Tang, Y. Synthesis of ZSM-48 zeolites and their catalytic performance in C4-olefin cracking reactions. Appl. Catal. A 2006, 299, 167–174. [Google Scholar]
- Wang, B; Gao, Q; Gao, JD; Ji, D; Wang, XL; Suo, JS. Synthesis, characterization and catalytic C4 alkene cracking properties of zeolite ZSM-23. Appl. Catal. A 2004, 290, 167–172. [Google Scholar]
- Zhu, XX; Liu, SL; Song, YQ; Xie, SJ; Xu, LY. Catalytic cracking of 1-butene to propene and ethene on MCM-22 zeolite. Appl. Catal. A 2005, 290, 191–199. [Google Scholar]
- Kaeding, WW; Chu, C; Young, LB; Weinstein, B; Butter, SA. Selective alkylation of toluene with methanol to produce para-Xylene. J. Catal 1981, 67, 159–174. [Google Scholar]
- Young, LB; Butter, SS; Kaeding, WW. Shape selective reactions with zeolite catalysts: III. Selectivity in xylene isomerization, toluene-methanol alkylation, and toluene disproportionation over ZSM-5 zeolite catalysts. J. Catal 1982, 76, 418–422. [Google Scholar]
- Chandawar, KH; Kulkarni, SB; Ratnasamy, P. Alkylation of benzene with ethanol over ZSM-5 zeolites. Appl. Catal. A 1982, 4, 287–295. [Google Scholar]
- Zhuang, JQ; Ma, D; Yang, G; Yan, ZM; Liu, XM; Liu, XC; Han, XW; Bao, XH; Xie, P; Liu, ZM. Solid-state MAS NMR studies on the hydrothermal stability of the zeolite catalysts for residual oil selective catalytic cracking. J. Catal 2004, 228, 234–242. [Google Scholar]
- Caeiro, G; Magnoux, P; Lopes, JM; Ramôa Ribeiro, F; Menezes, SMC; Costa, AF; Cerqueira, HS. Stabilization effect of phosphorus on steamed H-MFI zeolites. Appl. Catal. A 2006, 314, 160–171. [Google Scholar]
- Zhao, GL; Teng, JW; Xie, ZK; Jing, WQ; Yang, WM; Chen, QL; Tang, Y. Effect of phosphorus on HZSM-5 catalyst for C4-olefin cracking reactions to produce propylene. J. Catal 2007, 248, 29–37. [Google Scholar]
- Xue, NH; Chen, XK; Nie, L; Guo, XF; Ding, WP; Chen, Y; Gu, M; Xie, ZK. Understanding the enhancement of catalytic performance for olefin cracking: Hydrothermally stable acids in P/HZSM-5. J. Catal 2007, 248, 20–28. [Google Scholar]
- Kaeding, WW; Butter, SA. Production of chemicals from methanol: I. Low molecular weight olefins. J. Catal 1980, 61, 155–164. [Google Scholar]
- Vedrine, JC; Auroux, A; Dejaifve, P; Ducarme, V; Hoser, H; Zhou, S. Catalytic and physical properties of phosphorus-modified ZSM-5 zeolite. J Catal 1982, 73, 147–160. [Google Scholar]
- Lercher, JA; Rumplmayr, G. Controlled decrease of acid strength by orthophosphoric acid on ZSM-5. Appl. Catal 1986, 25, 215–222. [Google Scholar]
- Blasco, T; Corma, A; Martínez-Triguero, J. Hydrothermal stabilization of ZSM-5 catalytic-cracking additives by phosphorus addition. J. Catal 2006, 237, 267–277. [Google Scholar]
- Tynjala, P; Pakkanen, TT. Modification of ZSM-5 zeolite with trimethyl phosphite part 1. Structure and acidity. Micropor. Mesopor. Mater 1998, 20, 363–369. [Google Scholar]
- Xue, NH; Nie, Lei; Fang, DM; Guo, XF; Shen, JY; Ding, WP; Chen, Y. Synergistic effects of tungsten and phosphorus on catalytic cracking of butene to propene over HZSM-5. Appl. Catal. A 2009, 352, 87–94. [Google Scholar]
- Xu, J; Ying, JY. A Highly Active and Selective Nanocomposite Catalyst for C7+ Paraffin Isomerization. Angew. Chem. Int. Ed 2006, 45, 6700–6704. [Google Scholar]
- Rezgui, Y; Guemini, M. Effect of acidity and metal content on the activity and product selectivity for n-decane hydroisomerization and hydrocracking over nickel-tungsten supported on silica-alumina catalysts. Appl. Catal. A 2005, 282, 45–53. [Google Scholar]
- Di Gregorio, F; Keller, V. Activation and isomerization of hydrocarbons over WO3/ZrO2 catalysts: I. Preparation, characterization, and X-ray photoelectron spectroscopy studies. J. Catal 2004, 225, 45–55. [Google Scholar]
- Spamer, A; Dube, TI; Moodley, DJ; van Schalkwyk, C; Botha, JM. The reduction of isomerisation activity on a WO3/SiO2 metathesis catalyst. Appl. Catal. A 2003, 255, 153–167. [Google Scholar]
- Gielgens, LH; van Kampen, MGH; Broek, MM; van Hardeveld, R; Ponec, V. Skeletal Isomerization of 1-Butene on Tungsten Oxide Catalysts. J Catal 1995, 154, 201–207. [Google Scholar]
- Wang, QX; Zhang, SR; Cai, GY; Li, F; Xu, LY; Huang, ZX; Li, YY. Alkylation catalyst and the application thereof. US Patent 6,093,866 2000. [Google Scholar]
- Francesconi, MS; López, ZE; Uzcátegui, D; González, G; Hernández, JC; Uzcátegui, A; Loaiza, A; Imbert, FE. MFI/MEL intergrowth and its effect on n-decane cracking. Catal. Today 2005, 107, 809–815. [Google Scholar]
- Wang, P; Shen, BJ; Gao, JS. Synthesis of MAZ/ZSM-5 composite zeolite and its catalytic performance in FCC gasoline aromatization. Catal. Commun 2007, 8, 1161–1166. [Google Scholar]
- Fan, Y; Lei, D; Shi, G; Bao, XJ. Synthesis of ZSM-5/SAPO-11 composite and its application in FCC gasoline hydro-upgrading catalyst. Catal. Today 2006, 114, 388–396. [Google Scholar]
- Bouizi, Y; Rouleau, L; Valtchev, VP. Bi-phase MOR/MFI-type zeolite core–shell composite. Micropor. Mesopor. Mater 2006, 91, 70–77. [Google Scholar]
- González, G; González, CS; Stracke, W; Reichelt, R; García, L. New zeolite topologies based on intergrowths of FAU/EMT systems. Micropor. Mesopor. Mater 2007, 101, 30–42. [Google Scholar]
- Karlsson, A; Stöker, M; Schmidt, R. Composites of micro- and mesoporous materials: simultaneous syntheses of MFI/MCM-41 like phases by a mixed template approach. Micropor. Mesopor. Mater 1999, 27, 181–192. [Google Scholar]
- Solberg, SM; Kumar, D; Landry, CC. Synthesis, Structure, and Reactivity of a New Ti-Containing Microporous/Mesoporous Material. J. Phys. Chem. B 2005, 109, 24331–24337. [Google Scholar]
- Prokešov, P; Žilková, N; Mintova, S; Bein, T; Čejka, J. Catalytic activity of micro/mesoporous composites in toluene alkylation with propylene. Appl. Catal. A 2005, 281, 85–91. [Google Scholar]
- Kollár, M; Mihályi, RM; Pál-Borbély, G; Valyon, J. Micro/mesoporous aluminosilicate composites from zeolite MCM-22 precursor. Micropor. Mesopor. Mater 2007, 99, 37–46. [Google Scholar]
- Tan, QF; Bao, XJ; Song, TC; Fan, Y; Shi, G; Shen, BJ; Liu, CH; Gao, XH. Synthesis, characterization, and catalytic properties of hydrothermally stable macro-meso-micro-porous composite materials synthesized via in situ assembly of preformed zeolite Y nanoclusters on kaolin. J. Catal 2007, 251, 69–79. [Google Scholar]
- de Vos Burchart, E; Jansen, JC; van Bekkum, H. Ordered overgrowth of zeolite X onto crystals of zeolite A. Zeolites 1989, 9, 432–435. [Google Scholar]
- Millward, GR; Ramdas, S; Thomas, JM; Barlow, MT. Evidence for semi-regularly ordered sequences of mirror and inversion symmetry planes in ZSM-5/ZSM-11 shape-selective zeolitic catalysts. J.Chem. Soc. Faraday Trans 1983, 79, 1075–1082. [Google Scholar]
- Li, HX; Armor, JN. Low-silica EMT/FAU intergrowth zeolites with Si/Al = 1.0. Microporous Mater 1997, 9, 51–57. [Google Scholar]
- Goossens, AM; Wouters, BH; Grobet, PJ; Buschmann, V; Fiermans, L; Martens, JA. Synthesis and characterization of epitaxial FAU-on-EMT Zeolite overgrowth materials. Eur. J. Inorg. Chem 2001, 5, 1167–1181. [Google Scholar]
- Villaescusa, LA; Zhou, W; Morris, RE; Barrett, PA. Synthesis, characterization and control of faulting in STF/SFF topologies, a new family of intergrowth zeolites. J. Mater.Chem 2004, 14, 1982–1987. [Google Scholar]
- Zones, SI; Davis, ME. Zeolite materials: recent discoveries and future prospects. Curr. Opin Solid State Mater Chem 1996, 1, 107–117. [Google Scholar]
- Smirniotis, PG; Davydov, L; Ruchenstein, E. Composite Zeolite-Based Catalysts and Sorbents. Catal. Rev. Sci. Eng 1999, 41, 43–113. [Google Scholar]
- Kokotailo, GT. Crystalline zeolite product constituting ZSM-5/ZSM-11 intermediates. US Patent 4,229,424 1980. [Google Scholar]
- Kokotailo, GT. Catalytic conversion with crystalline zeolite product constituting ZSM-5/ZSM-11 intermediates. US Patent 4,289,607 1981. [Google Scholar]
- Dzkih, IP; Lopes, JM; Lemos, F; Ramoa Ribeiro, F. Mixing effect of USHY+HZSM-5 for different catalyst ratios on the n-heptane transformation. Appl. Catal. A 1999, 176, 239–250. [Google Scholar]
- Chen, HL; Shen, BJ; Pan, HF. In situ formation of ZSM-5 in NaY gel and characterization of ZSM-5/Y composite zeolite. Chem. Lett 2003, 32, 726–726. [Google Scholar]
- Han, Y; Xiao, FS. Catalytically active and hydrothermally stable mesoporous materials assembled from preformed nanosized zeolite precursors. Chin J Catal 2003, 24, 149–158. [Google Scholar]
- Niu, XL; Song, YQ; Xie, SJ; Liu, SL; Wang, QX; Xu, LY. Synthesis and catalytic reactivity of MCM-22/ZSM-35 composites for olefin aromatization. Catal. Lett 2005, 103, 211–218. [Google Scholar]
- Xie, SJ; Liu, SL; Liu, Y; Li, XJ; Zhang, WP; Xu, LY. Synthesis and characterization of MCM-49/ZSM-35 composite zeolites in the hexamethyleneimine and cyclohexamine system. Micropor. Mesopor. Mater 2009, 121, 166–172. [Google Scholar]
- Liu, Y; Zhang, WP; Xie, SJ; Xu, LY; Han, XW; Bao, XH. Probing the porosity of cocrystallized MCM-49/ZSM-35 Zeolites by hyperpolarized 129Xe NMR. J. Phys. Chem. B 2008, 112, 1226–1231. [Google Scholar]
- Qi, XL; Kong, DJ; Yuan, XH; Xu, ZQ; Wang, YD; Zheng, JL; Xie, ZK. Studies on the crystallization process of BEA/MOR co-crystalline Zeolite. J. Mater. Sci 2008, 43, 5626–5633. [Google Scholar]
- Qi, XL; Li, B; Li, SJ; Liu, XY; Lin, BX. Acidity and catalytic performance of BEA/MOR intergrowth zeolites. Chin J Catal 2006, 27(3), 228–232. [Google Scholar]
- Kong, DJ; Zheng, JL; Yuan, XH; Wang, YD; Fang, DY. Fabrication of core/shell structure via overgrowth of ZSM-5 layers on mordenite crystals. Micropor. Mesopor. Mater 2009, 119, 91–96. [Google Scholar]
- Tao, Y; Kanoh, H; Abrams, L; Kaneko, K. Mesopore-Modified zeolites: Preparation, characterization, and applications. Chem. Rev 2006, 106, 896–910. [Google Scholar]
- Hartmann, M. Hierarchical Zeolites: A proven strategy to combine shape selectivity with efficient mass transport. Angew. Chem., Int. Ed 2004, 43, 5880–5882. [Google Scholar]
- Christensen, CH; Johannsen, K; Schmidt, I; Christensen, CH. Catalytic benzene alkylation over mesoporous zeolite single crystals: improving activity and selectivity with a new family of porous materials. J. Am. Chem. Soc 2003, 125, 13370–13371. [Google Scholar]
- Lei, Q; Zhao, TB; Li, FY; Zhang, LL; Wang, Y. Catalytic cracking of large molecules over hierarchical zeolites. Chem. Commun 2006, 16, 1769–1771. [Google Scholar]
- Srivastava, R; Choi, M; Ryoo, R. Mesoporous materials with zeolite framework: Remarkable effect of the hierarchical structure for retardation of catalyst deactivation. Chem. Commun 2006, 43, 4489–4491. [Google Scholar]
- Kustova, MY; Hasselriis, P; Christensen, CH. Mesoporous MEL–type zeolite single crystal catalysts. Catal. Lett 2004, 96, 205–211. [Google Scholar]
- Schmidt, I; Krogh, A; Wienberg, K; Carlsson, A; Brorson, M; Jacobsen, CJH. Catalytic epoxidation of alkenes with hydrogen peroxide over first mesoporous titanium-containing zeolite. Chem. Commun 2000, 21, 2157–2158. [Google Scholar]
- Donk, SV; Janssen, AH; Bitter, JH; Jong, KPD. Generation, characterization, and impact of mesopores in zeolite catalysts. Catal. Rev 2003, 45, 297–319. [Google Scholar]
- Groen, JC; Zhu, WD; Brouwer, S; Huynink, SJ; Kapteijn, F; Moulijn, JA; Perez-Ramirez, J. Direct Demonstration of enhanced diffusion in mesoporous ZSM-5 zeolite obtained via controlled desilication. J. Am. Chem. Soc 2007, 129, 355–360. [Google Scholar]
- Christensen, CH; Johannsen, K; Törnqvist, E; Schmidt, I; Topsøe, H; Christensen, CH. Mesoporous zeolite single crystal catalysts: Diffusion and catalysis in hierarchical zeolites. Catal. Today 2007, 128, 117–122. [Google Scholar]
- Kloetstra, KR; Jansen, JC. Mesoporous material containing framework tectosilicate by pore–wall recrystallization. Chem Commun 1997, 2281–2282. [Google Scholar]
- Trong On, D; Kaliaguine, S. Ultrastable and highly acidic, zeolite-coated mesoporous aluminosilicates. Angew. Chem. Int. Ed 2002, 41, 1036–1040. [Google Scholar]
- Kirschhock, CEA; Kremer, SPB; Vermant, J; van Tendeloo, G; Jacobs, PA; Martens, JA. Design and synthesis of hierarchical materials from ordered zeolitic building units. Chem. Eur. J 2005, 11, 4306–4313. [Google Scholar]
- Egeblad, K; Christensen, CH; Kustova, M; Christensen, CH. Templating mesoporous zeolites. Chem. Mater 2008, 20, 946–960. [Google Scholar]
- Sato, K; Nishimura, Y; Shimada, H. Preparation and activity evaluation of Y zeolites with or without mesoporosity. Catal. Lett 1999, 60, 83–87. [Google Scholar]
- Benazzi, E; Lynch, J; Gola, A; Lacombe, S; Marcilly, C. Controlled removal of extra-framework aluminum species in USY Zeolite. In Proc 12th Int Zeolite Conf; 1999; Baltimore, USA; 5-10.07.1998; pp. 2735–2742.
- Groen, JC; Peffer, LAA; Moulijn, JA; Préz-Ramírez, J. Mechanism of hierarchical porosity development in MFI zeolites by desilication: the role of aluminium as a pore-directing agent. Chem. Eur. J 2005, 11, 4983–4994. [Google Scholar]
- Jacobsen, CJH; Madsen, C; Houzvicka, J; Schmidt, I; Carlsson, A. Mesoporous Zeolite Single Crystals. J. Am. Chem. Soc 2000, 122, 7116–7117. [Google Scholar]
- Schmidt, I; Boisen, A; Gustavsson, E; Stahl, K; Pehrson, S; Dahl, S; Carlsson, A; Jacobsen, CJH. Carbon nanotube templated growth of mesoporous zeolite single crystals. Chem. Mater 2001, 13, 4416–4418. [Google Scholar]
- Tao, Y; Kanoh, H; Kaneko, K. ZSM-5 monolith of uniform mesoporous channels. J. Am. Chem. Soc 2003, 125, 6044. [Google Scholar]
- Yang, Z; Xia, Y; Mokaya, R. Zeolite ZSM-5 with unique supermicropores synthesized using mesoporous carbon as a template. Adv. Mater 2004, 16, 727–732. [Google Scholar]
- Dong, AG; Wang, YJ; Tang, Y; Ren, N; Zhang, YH; Hong, YH; Gao, Z. Zeolitic tissue through wood cell templating. Adv. Mater 2002, 14, 926–929. [Google Scholar]
- Li, WC; Lu, AH; Palkovits, R; Schmidt, W; Spliethoff, B; Schuth, F. Hierarchically structured monolithic Silicalite-1 consisting of crystallized nanoparticles and its performance in the Beckmann Rearrangement of Cyclohexanone Oxime. J. Am. Chem. Soc 2005, 127, 12595–12600. [Google Scholar]
- Xiao, FX; Wang, LF; Yin, CY; Lin, KF; Di, Y; Li, JX; Xu, RR; Su, DS; Schlögl, R; Yokoi, T; Tatsumi, T. Catalytic properties of hierarchical mesoporous zeolites templated with a mixture of small organic ammonium salts and mesoscale cationic polymers. Angew. Chem., Int. Ed 2006, 45, 3090–3093. [Google Scholar]
- Wang, H; Pinnavaia, TJ. MFI Zeolite with small and uniform intracrystal mesopores. Angew. Chem. Int. Ed 2006, 45, 7603–7606. [Google Scholar]
- Choi, M; Cho, SH; Srivastava, R; Venkatesan, C; Choi, DH; Ryoo, R. Amphiphilic organosilane-directed synthesis of crystalline zeolite with tunable mesoporosity. Nature Mater 2006, 5, 718–723. [Google Scholar]
- Zhu, HB; Liu, ZC; Wang, YD; Kong, DJ; Yuan, XH; Xie, ZK. Nanosized CaCO3 as hard template for creation of intracrystal pores within Silicalite-1 crystal. Chem. Mater 2008, 20, 1134–1139. [Google Scholar]
- Liu, ZC; Kong, DJ; Wang, YD; Xie, ZK. Study on the mesoporous ZSM-5 syntheized with stach co-template. Acta Petrolei Sinica 2008, 24, 124. [Google Scholar]
- Zhu, HB; Liu, ZC; Wang, YD; Kong, DJ; Yuan, XH; Xie, ZK. Synthesis and catalytic performances of mesoporous zeolites templated by polyvinyl butyral gel as the mesopore directing agent. J. Phys. Chem. C 2008, 112, 17257–17264. [Google Scholar]
- Liu, Y; Zhang, WP; Liu, ZC; Xu, ST; Wang, YD; Xie, ZK; Han, XW; Bao, XH. Direct Observation of the mesopores in ZSM-5 zeolites with hierarchical porous structures by laser-hyperpolarized 129Xe NMR. J. Phys. Chem. C 2008, 112, 15375–15381. [Google Scholar]
- Hamilton, DJC. Homogeneous catalysis–new approaches to catalyst separation, recovery, and recycling. Science 2003, 229, 1702–1706. [Google Scholar]
- Li, C. Chiral Synthesis on catalysts immobilized in microporous and mesoporous materials. Catal. Rev. Sci. Eng 2004, 46, 419–492. [Google Scholar]
- McMorn, P; Hutchings, GJ. Heterogeneous enantioselective catalysts: strategies for the immobilisation of homogeneous catalysts. Chem. Soc. Rev 2004, 33, 108–122. [Google Scholar]
- Fan, QH; Li, YM; Chan, ASC. Recoverable catalysts for asymmetric organic synthesis. Chem. Rev 2002, 102, 3385–3466. [Google Scholar]
- Xiang, S; Zhang, Y; Xin, Q; Li, C. Asymmetric epoxidation of allyl alcohol on organic-inorganic hybrid chiral catalysts grafted onto the surface of silica and in the mesopores of MCM-41. Angew. Chem., Int. Ed 2002, 41, 821–824. [Google Scholar]
- Hu, A; Yee, GT; Lin, WB. Magnetically recoverable chiral catalysts immobilized on magnetite nanoparticles for asymmetric hydrogenation of aromatic ketones. J. Am. Chem. Soc 2005, 127, 12486–12487. [Google Scholar]
- Herron, N. A cobalt oxygen carrier in zeolite Y. A molecular “ship in a bottle”. Inorg. Chem 1986, 25, 4714–4717. [Google Scholar]
- Ogunwumi, SB; Bein, T. Intrazeolite assembly of a chiral manganese salen epoxidation catalyst. Chem. Commun 1997, 9, 901–902. [Google Scholar]
- Corma, A; Garcia, H. Supramolecular host-guest systems in zeolites prepared by ship-in-a-bottle synthesis. Eur. J. Inorg. Chem 2004, 6, 1143–1164. [Google Scholar]
- Dai, LX. Chiral Metal-Organic Assemblies–A new approach to immobilizing homogeneous asymmetric catalysts. Angew. Chem., Int. Ed 2004, 43, 5726–5729. [Google Scholar]
- Li, C; Zhang, HD; Jiang, DM; Yang, QH. Chiral catalysis in nanopores of mesoporous materials. Chem. Commun 2007, 6, 547–558. [Google Scholar]
- Diaz, JF; Balkus, KJ, Jr. Enzyme immobilization in MCM-41 molecular sieve. J. Mol. Catal. B: Enzym 1996, 2, 115–126. [Google Scholar]
- Yang, HQ; Li, J; Yang, J; Liu, ZM; Yang, QH; Li, C. Asymmetric reactions on chiral catalysts entrapped within a mesoporous cage. Chem. Commun 2007, 10, 1086–1088. [Google Scholar]
- Kim, TW; Ryoo, R; Kruk, M; Gierszal, KP; Jaroniec, M; Kamiya, S; Terasaki, O. Tailoring the pore structure of SBA-16 silica molecular sieve through the use of copolymer blends and control of synthesis temperature and time. J. Phys. Chem. B 2004, 108, 11480–11489. [Google Scholar]
- Kim, JM; Sakamoto, Y; Hwang, YK; Kwon, YU; Terasaki, O; Park, SE; Stucky, GD. Structural design of mesoporous silica by micelle-packing control using blends of amphiphilic block copolymers. J. Phys. Chem. B 2002, 106, 2552–2558. [Google Scholar]
- Yang, HQ; Zhang, L; Zhong, L; Yang, QH; Li, C. Enhanced cooperative activation effect in the hydrolytic kinetic resolution of epoxides on [Co(salen)] catalysts confined in nanocages. Angew. Chem. Int. Ed 2007, 46, 6861–6865. [Google Scholar]
- Hashiguci, S; Fujii, A; Takehara, J; Ikariya, T; Noyori, R. Asymmetric transfer hydrogenation of aromatic ketones catalyzed by Chiral Ruthenium (II) complexes. J. Am. Chem. Soc 1995, 117, 7562–7563. [Google Scholar]
- Liu, PN; Deng, JG; Tu, YQ; Gang, SH. Highly efficient and recyclable heterogeneous asymmetric transfer hydrogenation of ketones in water. Chem. Commun 2004, 18, 2070–2071. [Google Scholar]
- Inagaki, S; Guan, S; Fukushima, Y; Ohsuna, T; Terasaki, O. Novel mesoporous materials with a uniform distribution of organic groups and inorganic oxide in their frameworks. J. Am. Chem. Soc 1999, 121, 9611–9614. [Google Scholar]
- Asefa, T; MacLachlan, MJ; Coombs, N; Ozin, GA. Periodic mesoporous organosilicas with organic groups inside the channel walls. Nature 1999, 402, 867–871. [Google Scholar]
- Inagaki, S; Guan, S; Ohsuna, T; Terasaki, O. An ordered mesoporous organosilica hybrid material with a crystal-like wall structure. Nature 2002, 416, 304–307. [Google Scholar]
- Melde, BJ; Holland, BT; Blanford, CF; Stein, A. Mesoporous sieves with unified hybrid inorganic/organic frameworks. Chem. Mater 1999, 11, 3302–3308. [Google Scholar]
- Yoshina-Ishii, C; Asefa, T; Coombs, N; MacLachlan, MJ; Ozin, GA. Periodic mesoporous organosilicas, PMOs: fusion of organic and inorganic chemistry inside the channel walls of hexagonal mesoporous silica. Chem. Commun 1999, 24, 2539–2540. [Google Scholar]
- Asefa, T; Kruk, M; MacLachlan, MJ; Coombs, N; Grondey, H; Jaroniec, M; Ozin, GA. Novel bifunctional periodic mesoporous organosilicas, bpmos: synthesis, characterization, properties and in-situ selective hydroboration alcoholysis reactions of functional groups. J. Am. Chem. Soc 2001, 123, 8520–8530. [Google Scholar]
- Burleigh, MC; Markowitz, MA; Spector, MS; Gaber, BP. Direct synthesis of periodic mesoporous organosilicas: functional incorporation by co-condensation with organosilanes. J. Phys.Chem. B 2001, 105, 9935–9942. [Google Scholar]
- Burleigh, MC; Markowitz, MA; Spector, MS; Gaber, BP. Porous Organosilicas: An Acid-Catalyzed Approach. Langmuir 2001, 17, 7923–7928. [Google Scholar]
- Hamoudi, S; Kaliaguine, S. Periodic mesoporous organosilica from micellar oligomer template solution. Chem. Commun 2002, 18, 2118–2119. [Google Scholar]
- Sayari, A; Yang, Y. Nonionic oligomeric polymer directed synthesis of highly ordered large pore periodic mesoporous organosilica. Chem. Commun 2002, 21, 2582–2583. [Google Scholar]
- Das, D; Lee, JF; Cheng, S. Sulfonic acid functionalized mesoporous MCM-41 silica as a convenient catalyst for Bisphenol-A synthesis. Chem. Commun 2001, 21, 2178–2179. [Google Scholar]
- Dufaud, V; Davis, ME. Design of heterogeneous catalysts via multiple active site positioning in organic−inorganic hybrid materials. J. Am. Chem. Soc 2003, 125, 9403–9413. [Google Scholar]
- Díaz, I; Márquez-Alvarez, C; Mohino, F; Pérez-Pariente, J; Sastre, E. Combined alkyl and sulfonic acid functionalization of MCM-41-type silica: Part 2. Esterification of Glycerol with Fatty Acids. J. Catal 2000, 193, 295–302. [Google Scholar]
- Yang, QH; Kapoor, MP; Inagaki, S. Sulfuric acid-functionalized mesoporous benzene−silica with a molecular-scale periodicity in the walls. J. Am. Chem. Soc 2002, 124, 9694–9695. [Google Scholar]
- Yang, QH; Liu, J; Yang, J; Kapoor, MP; Inagaki, S; Li, C. Synthesis, characterization, and catalytic activity of sulfonic acid-functionalized periodic mesoporous organosilicas. J. Catal 2004, 228, 265–272. [Google Scholar]
- Li, CM; Yang, J; Shi, X; Liu, J; Yang, QH. Synthesis of SBA-15 type mesoporous organosilicas with diethylenebenzene in the framework and post-synthetic framework modification. Micropor. Mesopor. Mater 2007, 98, 220–226. [Google Scholar]
- Burleigh, MC; Markowitz, MA; Jayasundera, S; Spector, MS; Thomas, CW; Gaber, BP. Mechanical and hydrothermal stabilities of aged periodic mesoporous organosilicas. J. Phys. Chem 2003, 107, 12628–12634. [Google Scholar]
- Yang, Q; Li, Y; Zhang, L; Yang, J; Liu, J; Li, C. Hydrothermal stability and catalytic activity of aluminum-containing mesoporous ethane-silicas. J. Phys. Chem. B 2004, 108, 7934–7937. [Google Scholar]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar]
- Ajayan, PM; Zhou, OZ. Carbon Nanotubes: Synthesis, Structure, Properties, and Applications; Dresselhaus, MS, Dresselhaus, G, Avouris, Ph, Eds.; Springer-Verlag: Berlin, Germany, 2000; ; Chapter 13. [Google Scholar]
- Saito, R; Dresselhaus, G; Dresselhaus, MS. Physical Properties of Carbon Nanotubes; Imperial College Press: London, UK, 1999. [Google Scholar]
- Andrews, R; Weisenberger, MC. Carbon nanotube polymer composites. Curr. Opin. Solid State Mater. Sci 2004, 8, 31–37. [Google Scholar]
- Tasis, D; Tagmatarchis, N; Bianco, A; Prato, M. Chemistry of Carbon Nanotubes. Chem. Rev 2006, 106, 1105–1136. [Google Scholar]
- Sun, J; Gao, L; Li, W. Colloidal processing of carbon nanotube/alumina composites. Chem. Mater 2002, 14, 5169–5172. [Google Scholar]
- Ajayan, PM; Schadler, LS. Carbon nanotube filled polymer nanocomposites. Polym. Prep 2001, 42, 35–35. [Google Scholar]
- Barrera, E. Key methods for developing single-wall nanotube composites. JOM 2000, 52, 38–42. [Google Scholar]
- Biercuk, MJ; Llaguno, MC; Radosvljevic, M; Hyun, JK; Johnson, AT. Carbon nanotube composites for thermal management. Appl. Phys. Lett 2002, 80, 2767–2767. [Google Scholar]
- Ounaies, Z; Park, C; Wise, KE; Siochi, EJ; Harrison, JS. Electrical properties of single wall carbon nanotube reinforced polyimide composites. Compos. Sci. Technol 2003, 63, 1637–1646. [Google Scholar]
- Colbert, DT. Single-wall nanotubes: A new option for conductive plastics and engineering polymers. Plastics Add. Compd 2003, 3, 18–25. [Google Scholar]
- Sandler, JKW; Kirk, JE; Kinloch, IA; Shaffer, MSP; Windle, AH. Ultra-low electrical percolation threshold in carbon-nanotubeepoxy composites. Polymer 2003, 44, 5893–5899. [Google Scholar]
- Weisenberger, MC; Grulke, EA; Jacques, D; Rantell, T; Andrews, R. Enhanced mechanical properties of polyacrylonitrile/multiwall carbon nanotube composite fibers. J. Nanosci. Nanotech 2003, 3, 535–539. [Google Scholar]
- Maruyama, B; Alam, K. Carbon nanotubes and nanofibers in composite materials. SAMPE J 2002, 38, 59–70. [Google Scholar]
- Allaoui, A; Bai, S; Cheng, HM; Bai, JB. Mechanical and electrical properties of a MWNT/epoxy composite. Comp. Sci. Technol 2002, 62, 1993–1998. [Google Scholar]
- Jin, Z; Pramoda, KP; Xu, G; Goh, SH. Dynamic mechanical behavior of melt-processed multi-walled carbon nanotube/poly (methyl methacrylate) composites. Chem. Phys. Lett 2001, 337, 43–47. [Google Scholar]
- Vajtai, R; Wei, BQ; Zhang, ZJ; Jung, Y; Ramanath, G; Ajayan, PM. Building carbon nanotubes and their smart architectures. Smart Mater. Struct 2002, 11, 691–698. [Google Scholar]
- Sidney, A; Bernhard, EG; Louis, PH. Specific effects in acid catalysis by ion exchange resins. III. Some Observations on the Effect of Polyvalent Cations1. J. Am. Chem. Soc 1954, 76, 991–992. [Google Scholar]
- Strickler, GR; Lee, Guo-shuh J; Rierert, WJ. Process for the production of ethylene glycol. US Patent 9,825,985 1999. [Google Scholar]
- Shvets, VF; Kozlovskiy, RA; Kozlovskiy, IA; Makarov, MG; Suchkov, JP; Koustov, AV. The cause and quantitative description of catalyst deactivation in the ethylene oxide hydration process. Chem. Eng. J 2005, 107, 199–204. [Google Scholar]
- Li, YC; Yan, SR; Qian, LP; Yang, WM; Xie, ZK; Chen, QL; Yue, B; He, HY. Effect of tin on Nb2O5/α-Al2O3 catalyst for ethylene oxide hydration. J. Catal 2006, 241, 173–179. [Google Scholar]
- Van Hal, JW; Ledford, JS; Zhang, XK. Investigation of three types of catalysts for the hydration of ethylene oxide (EO) to monoethylene glycol (MEG). Catal. Today 2007, 123, 310–315. [Google Scholar]
- Yu, FP; Cai, H; He, WJ; Yang, WM; Xie, ZK. Synthesis and characterization of a polymer/multiwalled carbon nanotube composite and its application in the hydration of ethylene oxide. J. Appl. Polymer Sci 2009, 115, 2946–2954. [Google Scholar]












© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xie, Z.; Liu, Z.; Wang, Y.; Yang, Q.; Xu, L.; Ding, W. An Overview of Recent Development in Composite Catalysts from Porous Materials for Various Reactions and Processes. Int. J. Mol. Sci. 2010, 11, 2152-2187. https://doi.org/10.3390/ijms11052152
Xie Z, Liu Z, Wang Y, Yang Q, Xu L, Ding W. An Overview of Recent Development in Composite Catalysts from Porous Materials for Various Reactions and Processes. International Journal of Molecular Sciences. 2010; 11(5):2152-2187. https://doi.org/10.3390/ijms11052152
Chicago/Turabian StyleXie, Zaiku, Zhicheng Liu, Yangdong Wang, Qihua Yang, Longya Xu, and Weiping Ding. 2010. "An Overview of Recent Development in Composite Catalysts from Porous Materials for Various Reactions and Processes" International Journal of Molecular Sciences 11, no. 5: 2152-2187. https://doi.org/10.3390/ijms11052152
APA StyleXie, Z., Liu, Z., Wang, Y., Yang, Q., Xu, L., & Ding, W. (2010). An Overview of Recent Development in Composite Catalysts from Porous Materials for Various Reactions and Processes. International Journal of Molecular Sciences, 11(5), 2152-2187. https://doi.org/10.3390/ijms11052152