From the standpoint of information theory, any signal is a physical structure for which temporal or spatial changes of state encode information, or recognized contrast with the environment. In the neuron, signals include ligand- and voltage-gated ion channels, G-protein-coupled receptors, receptor tyrosine kinases, and a variety of other molecular factors. The simplest function of a microdomain is to provide a scaffold or platform to co-localize the molecular signals, thus facilitating their interaction [
24–
26]. For example, a neuron receptor could be localized only to those microdomains which contain a specific set of signal components, thereby preventing or significantly limiting non-specific signaling. A more complex possibility exists: In a microdomain-based switching sytem, the components necessary to activate or modulate a signaling pathway could initially be segregated into separate microdomains. Subsequent microdomain fusion, induced by an external input, could activate or refine the signal. The separation of the microdomains, following the termination of the input, would then re-set the system. It is important to note that in both types of processes, microdomains are essentially passive, providing platforms for proteins. Is it possible that microdomains may also play a more active role, modulating by means of their intrinsic chemical structure intracellular traffic and electrical signaling in the neuron? In this section, we review recent studies of neuron microdomains, and evaluate this possibility.
4.1. Neural Microdomains, Development, and Plasticity
Viewed from an adaptive standpoint, the highly dynamic environments of the more behaviorally complex species (especially social carnivores, nonhuman primates, and humans) require constant structural changes in their information-processing machinery. As a result, many—if not all—of the intracellular molecular systems which regulate changes in neural structure and network connectivity from embryo to maturity continue to operate throughout adult life, thereby blurring the conventional distinction between development and plasticity. Moreover, because some of these systems mediate between electrical or molecular inputs and the information encoded in genes, they are of considerable theoretical importance in the biology of human behavior [
27]. Increasing evidence suggests that microdomains may play a major role in activating or modulating these pathways. One striking example is neurotrophic-factor, or neurotrophin, signaling.
Neurotrophins are polypeptides critical to the growth, differentiation and survival of immature neurons, as well as to the maintenance of neurons in the adult nervous system. One of the best-understood neurotrophins in neuron growth factor (NGF), extensively studied for sixty years. Historically, NGF played a significant role in Victor Hamburger and Rita Levi-Montalcini’s formulation of the Neurtrophic Factor Hypothesis (NFH) [
28,
29]. The investigators demonstrated that the size of a developing neuron could be modified by changing the size of its innervation target. They explained this result by proposing a “metabolic exchange between the neurite and the substrate to which it grows”. Implicit in NFH was the concept that neurotrophins, synthesized and secreted by the target neuron, bind with receptors on the innervating neuron in a form of retrograde signaling. Subsequent studies of NGF and the other neurotrophins, Brain-Derived Neurotrophic Factor (BDNF), NT-3, NT-4/5, and NT-6, have strongly supported NFH [
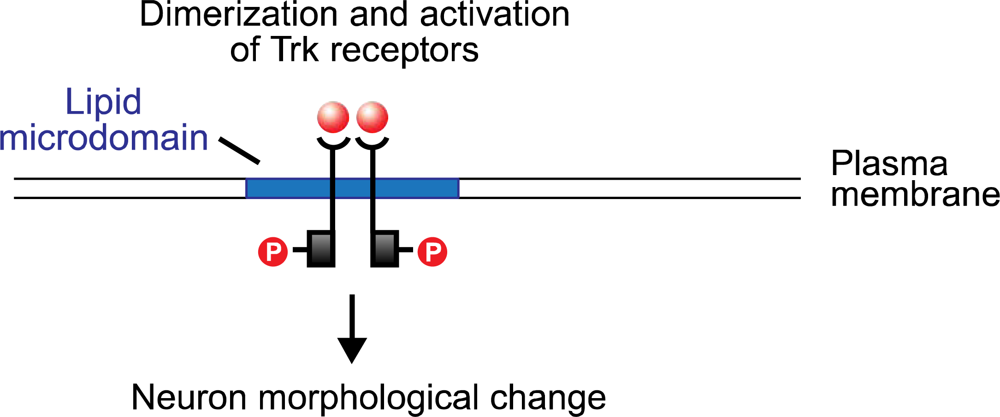
30]. The neurotrophins bind to the tyrosine kinase (Trk) receptors TrkA, TrkB, and TrkC, and to the low-affinity glycoprotein receptor p75. (The last, a tumor necrosis factor, is structurally unrelated to the Trk proteins, and appears to potentiate the pathway activated by NGF binding with TrkA.) The neurotrophin molecule binds as a dimer, and in turn causes the dimerization of the Trk receptor (
Figure 2). The cytoplasmic tyrosine kinase domains on the polypeptide chains of the Trk dimer then cross-phosphorylate, creating docking sites for the src homology domain 2 (SH-2), present on several cellular proteins. One of these is Shc which, following binding with the phosphotyrosine region of the Trk receptor, is itself phosphorylated by Trk. Shc binding and activation initiates a cascade of phosphorylation events, the first stage of which terminates with the exchange of GDP for GTP on the ras protein. Ras, which is active in its GTP-bound form, initiates a second phosphorylation sequence terminating in the activation of the Extracellular Response Kinase (ERK) protein. ERK initiates two pathways, both of which are significant to neuron modification: one is a phosphorylation sequence involving numerous cellular proteins including tyrosine hydroxylase and microtubule proteins; the other is the entry of activated ERK into the nucleus where it interacts with transcription factors that regulate the rate of mRNA synthesis.
Although much is still unknown regarding the final stages of neurotrophin-induced signaling—e.g., the precise sequences that link ERK activation with neuron morphological change—two conclusions may be reached regarding the neurotrophin-Ras-ERK pathway. One is that microdomains are clearly crucial for effective neurotrophin signaling [
31–
33]. As noted above, a fundamental function of the microdomain is a scaffold or platform which co-localizes molecular signals, thereby facilitating their interaction. TrkA and B are enriched in microdomains, which enhances autophosphorylation following neurotrophin binding. Following cholesterol depletion with cyclodextrin, Trk autophosphorylation is disrupted by >50% (but see the cautionary discussion above). Also, many intermediates in the Trk cascade are microdomain-associated, including Shc and ras. Perhaps the latter finding exemplifies the separation of signal components into separate microdomains when the initial signaling molecule (Trk) is not activated, followed by microdomain fusion after neurotrophin binding. This, and other features of microdomain-modulated intracellular signaling, requires further investigation. The second conclusion that may be drawn is that neurotrophin signaling occurs in the adult brain, thus illustrating the fading distinction between development and plasticity. James Conner’s team recently demonstrated that NGF infusions into the adult rat hippocampus enhanced long-term potentiation (LTP), an electrophysiological correlate of neural plasticity [
34]. Conversely, NGF blockade through infusion of an NGF antibody, significantly reduced LTP and impaired spatial learning. In view of separate evidence that other cognitive functions, including novelty detection, are localized in the hippocampus, it would be of interest to determine if these were modifiable through NGF augmentation and blockade.
In contrast to neurotrophin signaling, the molecular sequences involved in axon elongation and targeting are less well understood. There is as yet only limited evidence suggesting that the response of the neural growth cone, the region at the leading tip of the neurite, to an extracellular diffusion gradient of molecules produced and released by a target neuron may be microdomain-mediated. Growth-cone navigation is initiated by the binding of the extracellular cues (comprising the diffusion gradient) to plasma membrane receptors, followed by receptor oligomerization and complex formation with co-receptors or other signal proteins [
35–
37] Plasma membrane complex formation in turn stimulates an intracellular cascade resulting in cytoskeleton re-organization which transforms neurite morphology. The precise targeting which is essential for constructing a neural network is achieved by spatial asymmetry of the plasma membrane signaling complexes. Anterograde migration is generated by a shift in receptor activation toward the front of the growing cell, while neurite turning results from shifts in activation to the left or right. These changes are in turn converted into the activation and inhibition of intracellular signaling pathways which ultimately regulate navigation. A few key studies suggest that microdomains may be essential scaffolds for concentrating the signaling molecules. In a recent set of experiments conducted by Carmine Guirland’s team, microdomains in developing neurons were selectively disrupted by the application of methyl-β-cyclodextrin (MCD) which depletes membrane cholesterol (see earlier methodological discussion), and the effect on navigation was monitored by light microscopy [
38]. As a precaution against changes in neurite mobility being an artifact of chlolesterol depletion, microdomains were also disrupted using filipin, which disrupts microdomains without extracting cholesterol. The results strongly suggested an important role for microdomains in neuron development. Neuron responses to diffusion gradients of several extracellular cues, including BDNF, neutrin-1, and Semaphorin 3A, were blocked by disrupting microdomain integrity. In a similar vein, Hiroyuki Kamiguchi’s group inactivated microdomains by fluorescein-labeling a GMI ganglioside (contained in the microdomain outer leaflet) which in turn releases singlet oxygen upon laser irradiation [
39]. The disruptive effect of the singlet oxygen is highly focal, extending for only 5 nm. Consistent with Guirland’s results, the response of the growth cone tip to N-cadherin and L1, two cell adhesion molecules (CAMs) was blocked by microdomain disruption. Together these data suggest that, while many features of the process are still imperfectly understood, it is becoming evident that microdomains are essential to axon targeting during brain development. Important questions for future research include the identification of the specific intracellular pathways involved, the mechanics of pathway cross-talk, and the extent to which these cascades play a role in adult plasticity.
In pursuing the latter questions, the relatively well-defined interactions involved in activity-dependent signaling to the nucleus via cyclic AMP response element binding protein (CREB) may possibly serve as a prototype. Current models of the CREB pathway specify, to a relatively complete extent, a regulatory system that begins with cystolic Ca
2+ elevation, extends to CREB phosphorylation, and terminates with evolutionarily-related behaviors such as spatial mapping, fear conditioning, and circadian activity [
40–
42]. The system is therefore important as a paradigmatic example of gene-brain reciprocal signaling in the biology of human behavior [
43.
44]. Cholesterol-depletion experiments which disrupt downstream pathways (but see earlier cautionary remarks regarding this method) suggest that Ca
2+ entry into the cytosol, which initiates the CREB cascades may be stabilized by microdomain association and clustering of signal components. L-type voltage-sensitive Ca
2+ channels (L-VSCCs), the ligand-gated Ca
2+-conducting N-methyl-D-aspartate (NMDA) glutamate receptor channel (co-localized wth the Ca
2+-conducting α-amino-3-hydroxy-5-methyl-4- isoxazolepropionic (AMPA) channel), and the epidermal growth factor (EGF) tyrosine kinase receptor involved in Ca
2+ release, are all microdomain-localized. Biswaranjan Pani and Birj Singh, in a recent review [
45], propose that microdomains stabilize these signal components, a view consistent with Kusumi’s oligomerization-induced trapping model discussed above. This view appears particularly plausible in the relation to the NMDA and AMPA receptor channels, in that intensive glutamate activation of AMPA must coincide with glutamate binding with NMDA to remove NMDA channel blockade by extracellular magnesium, permitting Ca
2+ influx. This “coincidence detector” function would likely not be possible if the receptors were not co-localized by means of microdomain stabilization. An alternative modality is Ca
2+ release from endoplasmic reticulum (ER) stores. Hormone or growth-factor binding with microdomain-associated tyrosine kinase receptors initiates a signal cascade which generates inositol 1,4,5 triphosphate (IP
3). IP
3 in turn binds with ER-localized IP
3 receptors, accomplishing Ca
2+ release from the intracellular stores. In an intriguing—and pioneering—discussion, Pani and Singh propose that co-localization of the Ca
2+-conducting ER TRPC channels and the ER STIM1 proteins which regulate them, is facilitated by ER microdomain scaffolding. If their model turns out to be correct, it would suggest that microdomain localization is essential for spatio-temporal coordination of protein dynamics on both the organelle and plasma-membrane levels.
The series of events following Ca
2+ elevation includes several mechanisms for Ca
2+-CREB-gene signal amplification and damping. (An important, if daunting, question for future research is the relative strength of each mechanism’s contribution to the final behavioral phenotype.) A critical early event is Ca
2+ binding with the calmodulin protein (CaM). The latter may be regarded as a node in a molecular network which, given its multiple targets, could have an amplifying effect. CaM targets the protein kinases CaMKI, CaMKII, and especially CaMKIV, each of which phosphorylates CREB at a serine site (Ser-133), an event required for CREB function. The CREB protein, which is pre-bound to the CREB response element (CRE) promoter, is then enabled to recruit the CREB-binding protein (CBP) co-activator, which generates transcription activity. In addition to the Ca
2+ cascade, CREB is activated through other important pathways including those initiated by neurotrophin binding with Trk receptors (see earlier discussion). Cross-talk between these pathways is another probable means by which input to CREB, and consequent up- or down-regulation of gene transcription, may be rapidly effected. A final modulatory mechanism which has only recently come to light involves modification of both CREB and CBP following Ser-133 phosphorylation and CBP recruitment. At this stage of the cascade, CREB may be additionally phosphorylated at Ser-142 and Ser-143 sites while CBP may be phosphorylated at Ser-301, and methylated at an arginine residue. As Bonnie Lonze and David Ginty have suggested, it is plausible that maximal phosphorylation may be associated with maximal gene expression [
46]. Building upon that premise, the various modulatory mechanisms appear consistent with the concept of gene-brain cross-talk facilitating adaptation in complex, changing environments. Perhaps not surprisingly, brain systems currently identified with Ca
2+-CREB-gene signaling are neural substrates for ancient behaviors—circadian activity, spatial mapping, fear conditioning, memory of defeat in aggressive encounters—for which environmentally-sensitive plasticity coupled with genome stabilization would have been essential for species survival. A full discussion of these systems is beyond the scope of this review. However, we may note that the final impression one takes away from these intricate, linked, and graded intracellular pathways is that of a hierarchy of evolutionarily optimized molecular regulators, at the apex of which is the neural membrane microdomain.
4.2. Microdomains, Action Potential Propagation, and Synaptic Transmission
In contrast to the intensive research on microdomain involvement in intracellular signaling, there has been relatively less emphasis on the possible role of these structures in impulse propagation. The disparity is somewhat surprising, given the strong historical priority of research on the latter topic. According to the conventional model formally described in 1952 by Alan Lloyd Hodgkin and Andrew Huxley, and based on their experiments with the squid giant axon, the neural impulse or action potential (AP) is initiated and propagated by means of trans-membrane ion flux through protein channels which successively generates, along the axon, a reversal of membrane potential at each channel locus above a critical threshold, thus relaying the impulse [
47]. Nearly six decades later, the Hodgkin-Huxley (HH) model, although subjected to occasional critiques, remains the widely accepted explanation for electrical signaling in the neuron. Moreover, it has strongly influenced formal mathematical modeling of the artificial neuron, to be discussed briefly in the following section. Although microdomain stabilization of ion channels and channel co-factors is clearly relevant to HH, there is at the time of this writing only one major study of microdomain modulation of ion-channel properties. Jeffrey Martens’ team investigated the effect of cholesterol depletion on the electrical properties of the microdomain-associated potassium channel Kv2.1 [
48,
49]. The study determined that cholesterol depletion by cyclodextrin caused a ∼36 mV hyperpolarizing shift (from 15.7 [control] to −51.6 [cyclodextrin-treated]) in the midpoint of the Kv2.1 inactivation curve. Importantly, cyclodextrin treatment of the non-microdomain-associated channel Kv4.2 showed no observable effect on channel properties. The latter finding suggests that the modulation of Kv2.1 inactivation was due to the effect of cyclodextrin on the channel’s microdomain environment (including microdomain-associated signal proteins such as Trk), and not to damage to other cellular structures (e.g., the cytoskeleton). The study notes that Kv channels often contain phosphorylation sites, and that Kv2.1 in particular is tyrosine-phosphorylated. The observed inactivation effect may thus have resulted from cyclodextrin-induced separation of scaffolded signal components rather than from disruption of direct-microdomain-lipid interaction with the ion-channel protein.
The Martens experiment, like most of the studies in this review, emphasizes the scaffolding function of microdomains. Are there also intrinsic microdomain physical and chemical properties that modulate the activity of neural-membrane integral proteins, including in particular ion channels? To date, the answer is largely conjectural, suggesting a need for further research. Computer-simulation studies conducted by Harry Price and the author suggest that electric-field effects generated in the plane of the membrane during AP propagation may transiently alter microdomain physical properties in a manner that could modulate ion-channel activity, thereby altering neuron signaling [
50,
51]. Studies conducted with both natural and artificial membranes indicate that an applied electric field can produce electrophoretic movement of membrane proteins, and re-organize the bilayer [
52–
54] Concanavalin A (conA) protein receptors in living cells were redistributed in response to a 4 V/cm electric field, and phase separation of a two-component supported membrane was observed following the application of a 10–30 V/cm field. These data, and the results of similar experiments, are to be compared with the field strength of 100 kV/cm generated in the membrane at the peak of the AP.
On the basis of these and similar studies, we simulated the possible mechanical and electrostatic effects of an applied field on different membrane-lipid species. In our initial study, we investigated how the alignment of unsaturated bonds of model compounds would affect field-responsive properties (dipole and quadrupole moments, and polarizability). The results of our first model indicated a strongly increased sensitivity to field gradients as the number of unsaturated bonds is increased. In relation to this finding, a second, more complex, study investigated energetically-favorable clustering, and polarizability values, of model lipids (including, importantly, sphingolipids). It was found that sphingolipids display an energetically-favorable alignment of unsaturated bonds. (In an actual neuron, the alignment would be in the plane of the bilayer). In addition, the aligned bonds of the sphingolipids displayed the highest polarizability of all model lipid clusters. We then intuitively proposed that, in a neuron, transient dipoles would be generated in the clustered sphingolipids of an ion-channel associated microdomain as a field effect of channel gating and the trans-membrane current. We further suggested that the lipid dipoles would interact electrostatically with charge residues known to be present in an unfolded (random coil) ion-channel protein. Ion-channel closing (
i.e., a return to the α-helix conformation) could therefore be regarded as many-particle system seeking a minimum local potential energy. Put differently, the duration of the ion-channel “open” state, and hence the membrane potential, would be a function of the duration of lipid-protein electrostatic interactions in an ion-channel-associated microdomain. It would be, of course, incautious to infer too much from a limited number of studies. But the data appear to suggest that transient electrostatic interactions between a microdomain’s sphingolipids and an associated, voltage-gated, ion-channel protein may modulate AP propagation. Recalling Martens’ data, one might envision a variation of Stephen Waxman’s “multiplex” neuron in which an array of communicating modules with distinctive physical properties (electrostatics, phosphorylation) play specialized informational roles [
55]. This possibility will be examined in more detail in the discussion of artificial intelligence.
The final stage of AP propagation is the relay of impulses into the neuron terminal, an event which generates intricate but temporally- and spatially-coordinated synaptic-transmission processes that are the basis of inter-neural signaling. In outline, synaptic transmission consists of the exocytosis of transmitter substance from lipid vesicles transiently fused with the pre-synaptic-terminal plasma membrane; the exocytosed neurotransmitter binds with post-synaptic receptors, resulting in target membrane depolarization, and continued AP propagation. Increasing evidence suggests that certain features of exocytosis may be microdomain-regulated [
56–
58]. It is important to note, however, that several aspects of the process are not well understood, and the richness of scenarios has occasionally surpassed that of the data. Given this caveat, it is clear that transmitter release is initiated by the influx of Ca
2+ ions through microdomain-associated, voltage-gated channels in the presynaptic terminal plasma membrane [
59]. Calcium ion elevation accomplishes the translocation of vesicles from an interior reserve pool to a “readily-releasable pool” (RRP) in the active zone of the pre-synaptic terminal, where they are docked and primed before fusing with the plasma membrane and releasing their transmitter cargo. Vesicle movement from the interior pool to the RRP is mediated by synapsin. This phosphoprotein is vesicle-associated and bound to actin; the latter may provide a scaffold for interior-reserve vesicles. Synapsin is phosphorylated by several different kinases (including CaMKII and protein kinases A and C), which causes it to dissociate from the interior reserve pool and translocate to the active zone of the presynaptic terminal plasma membrane (docking stage). It is estimated that ∼10–30 vesicles are docked at any one time, and that the majority are incapable of transmitter release;
i.e., the docked vesicles require maturation, or priming. The steps involved in the latter process remain somewhat undefined, but it is possible that the vesicles may partially fuse with the plasma membrane, and that the energy derived from ATP hydrolysis may alter the conformation of proteins comprising the exocytic machinery. Fusion, the following stage, is triggered by Ca
2+ ions binding with negatively-charged aspartate residues (C2A and C2B) in a vesicle-associated protein sensor, synaptotagmin. Synaptotagmin may undergo an electrostatic rather than conformational change, causing it to interact with an elaborate vesicle-and plasma-membrane-associated protein machinery—the much-analyzed SNARE complex—that directly induces membrane fusion and transmitter exocytosis.
On purely intuitive grounds, it would seem that SNARE complex function would require microdomain localization of its protein components for rapid assembly, coordinated function, and disassembly. But artificial-membrane and
in vivo membrane data are highly inconsistent, contrasting noticeably with the greater convergence in the SNARE-protein imaging studies [
60–
62]. SNARE (soluble N-ethylmaleimide-sensitive fusion protein (NSF) attachment protein (SNAP) receptor) association with microdomains has been documented for Madin-Darby canine kidney cells, 323-L1 adipocytes, HeLa cells, and rat brain somatosomes. However, artificial-membrane studies of the SNARE components syntaxin 1 (plasma membrane-associated SNARE) and synaptobrevin 2/VAMP2 (vesicle-associated SNARE) indicated a preference for the liquid-disordered phase. Interpretations of microdomain modulation of exocytosis are thus an uneasy blend of consensus and speculation. Based on X-ray crystallographic, FRET, and atomic force microscopy (AFM) studies, it is clear that the SNARE complex is formed by a zippering process, proceeding from N- to C-termini of the SNARE domains, in which the SNARE vesicle component synaptobrevin 2/VAMP2 is progressively tethered to the plasma-membrane SNARE components syntaxin and SNAP-25. Importantly, zippering/tethering is only possible when the Munc-18 protein separates from syntaxin, indicating a switching function. The role of microdomains in subsequent vesicle and plasma membrane fusion has been the subject of much speculation. A provocative set of models proposed by Christine Salaün’s team attempts to reconcile some of the conflicting data by proposing various ways in which microdomains
and liquid-disordered domains would segregate different types of exocytosis [
63]. For example, liquid-disordered domains would be the site of “kiss-and-run” exocytosis, in which a partial fusion of vesicle and plasma membranes creates a transient aqueous pore through which transmitter is released; alternatively, microdomains would be the site for full fusion of the two membranes. In what is clearly a programmatic article, the exact opposite scenario is also proposed, and other models are suggested as well. When exocytosis is complete—by means of one or several of these mechanisms—the NSF protein binds with SNAP-25 and, via energy derived from ATP hydrolysis, dissociates the SNARE complex for another exocytic cycle.
Exocytosed transmitter substances bind with a wide variety of post-synaptic receptors. It is becoming increasingly clear that microdomain and liquid-disordered membrane regions help regulate receptor function by alternately segregating and co-localizing receptor signal components; moreover, microdomains may also play an active role in receptor conformational changes essential for transmitter binding [
64–
67]. These functions have been investigated in a number of prototype systems, especially AMPA, acetylcholine, and serotonin. The AMPA receptor, as noted briefly above, functions together with the NMDA receptor in a “coincidence detector” mechanism; their interaction is a molecular substrate for long-term-potentiation (LTP), the neurophysiological basis for learning. A full discussion of LTP’s molecular pathways is beyond the scope of this review. Essentially, the AMPA and NMDA receptors are activated by glutamate, the most abundant neurotransmitter in the central nervous system. Glutamate binds with both receptor types, but opens only the AMPA receptor (causing Na
+ influx) due to Mg
2+ occlusion of the NMDA receptor channel pore. Subsequent depolarization of the AMPA receptor causes the repulsion of Mg
2+, allowing Ca
2+ influx. Calcium entry in turn generates a cascade of LTP-associated events. These include the upregulation of AMPA receptors to the membrane, and AMPA receptor phosphorylation by CaMKII, which increases its channel conductance. Recently, Qingming Hou’s team demonstrated microdomain localization of AMPA receptors by fluorescence imaging of double-labeled AMPA receptors and microdomains in cultured hippocampal neurons [
68]. Additionally, their study determined that microdomain localization of AMPA receptors in cultured cortical neurons was increased by NMDA receptor activity, an effect which was abolished by an NMDA antagonist. The authors of the study propose that NMDA receptor activity may recruit AMPA receptors to microdomains, although the mechanisms remain uncertain. Once recruited, AMPA receptors may be phosphorylated by microdomain-associated Trk, possibly altering AMPA receptor activity levels and thereby modulating synaptic plasticity.
Hou’s research strongly suggests a microdomain scaffolding function which accomplishes the co-localization of AMPA and NMDA receptor signaling components. But is it possible that microdomains affect receptor dynamics in a more direct way? Jacques Fantini and Francisco Barrantes, in a recent review of receptor-microdomain interactions [
69], propose that cholesterol and sphingolipids “are active partners of synaptic transmission.” Their model bridges several orders of magnitude, linking the physical chemistry of the acetylcholine and serotonin receptors with the regulation of transmitter binding and signal transduction. In the case of the acetylcholine receptor, a wide variety of imaging studies indicate that its microdomain environment is subdivided into bulk and shell lipids, both in a liquid-ordered phase. The shell component immediately surrounds the receptor, restricting its mobility for possible protein-cholesterol interaction. Although a cholesterol-binding motif has not yet been clearly identified for the acetylcholine receptor, several studies have shown that the receptor’s ordered α-helical structure and, importantly, ligand-binding stability, are both cholesterol-dependent. Sphingolipids may also be essential to effective acetylcholine-receptor function. Structural studies of a wide range of proteins indicate a sphingolipid binding domain (SBD), comprised of amino acid residues which bind to the polar headgroup of the sphingolipid. Based on these data, the investigators propose a ternary complex in which “cholesterol interacts simultaneously with one of the transmembrane domains of the receptor and with a glycosphingolipid.” The conformation of each of the three would be influenced by their cross-talk, a dynamic which in turn may regulate receptor sensitivity to neurotransmitter binding. Similarly, ligand binding and intracellular signaling of the G protein-coupled serotonin receptor depends upon microdomain integrity; receptor functions are disrupted by sphingolipid and/or cholesterol depletion, and recovered by its restoration. Based upon these data, and recent infrared and ultraviolet spectroscopy evidence for eight low-energy conformational isomers of serotonin [
70], the investigators propose that interaction between cholesterol, sphingolipids, and the G protein-coupled serotonin receptor may regulate ligand binding and receptor-mediated signaling. In effect, the interactions within the ternary complex would “mold” the highly flexible serotonin molecule into an isomer specific for a serotonin-receptor subset. Microdomain sphingolipids, which display remarkable conformational variety (numbering in the hundreds of shapes) due to chain length, degree of saturation, and head-group structure, would play a determinative role in fashioning a distinctive shape for the serotonin molecule.
The computational implications of the Fantini-Barrantes model are worth considering. In essence, their viewpoint appears consistent with a concept of sub-neural information-processing modules in which the tokens are a set of interacting molecules seeking a minimum local potential energy. Receptor-channel gating and subsequent field-induced lateral movement of sphingolipids between post-synaptic microdomains, possibly by hop diffusion over cytoskeleton barriers, would modulate the computation, thus amounting to a form of learning. Possibilities such as these reflect the growing conceptual richness of microdomain theory, now entering its third decade. They also suggest a role for molecular-machine mimics in extending current models of artificial intelligence. This strategy will be discussed briefly in the following section.

{kind=link}
{kind=link}
{kind=link}


