Bioengineering Embryonic Stem Cell Microenvironments for the Study of Breast Cancer


Abstract
:
1. Introduction
2. Characteristics of Breast Cancer Cells and Tumor Microenvironments
2.1. Uncontrolled Tumor Growth
2.2. Metastasis
2.3. Tumor Microenvironment
2.4. Reprogramming Breast Cancer Cells
3. Characteristics of Embryonic Stem Cells and Microenvironments
3.1. Self-Renewal and Pluripotency of Embryonic Stem Cells
3.2. Embryonic Microenvironments
3.3. Bioengineering Embryonic Stem Cell Microenvironments
4. Embryonic Microenvironment and Cancer
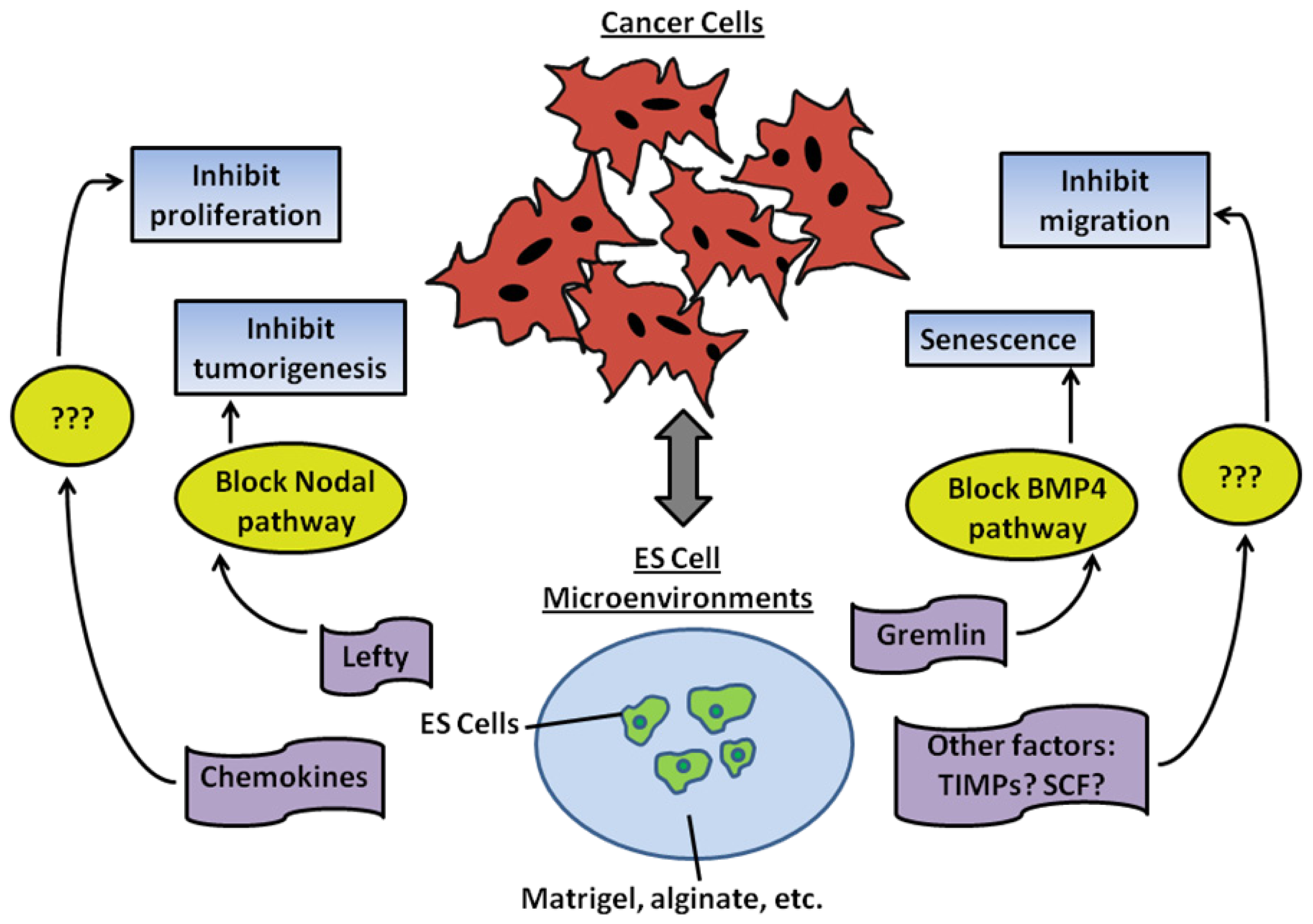
4.1. The Importance of Embryonic Microenvironments in Inhibiting Tumorigenesis
4.2. Convergence of Tumorigenic and Embryonic Signaling Pathways
4.3. Breast Cancer Stem Cells and Embryonic Stem Cell Microenvironment
4.4. Reprogramming of Metastatic Cancer Cells Using ES Cell-Conditioned Microenvironments
4.5. Bioengineered Embryonic Microenvironments for Breast Cancer Research
5. Outlook
Acknowledgements
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin 2011, 61, 212–236. [Google Scholar]
- Gluck, S. The prevention and management of distant metastases in women with breast cancer. Cancer Invest 2007, 25, 6–13. [Google Scholar]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar]
- Finger, E.C.; Giaccia, A.J. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev 2010, 29, 285–293. [Google Scholar]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar]
- Bissell, M.J.; Labarge, M.A. Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell 2005, 7, 17–23. [Google Scholar]
- Weigelt, B.; Bissell, M.J. Unraveling the microenvironmental influences on the normal mammary gland and breast cancer. Semin. Cancer Biol 2008, 18, 311–321. [Google Scholar]
- Ma, X.J.; Dahiya, S.; Richardson, E.; Erlander, M.; Sgroi, D.C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 2009, 11, R7:1–R7:18. [Google Scholar]
- Carlini, M.J.; de Lorenzo, M.S.; Puricelli, L. Cross-talk between tumor cells and the microenvironment at the metastatic niche. Curr. Pharm. Biotechnol 2011, in press. [Google Scholar]
- Croci, D.; Salatino, M. Tumor immune escape mechanisms that operate during metastasis. Curr. Pharm. Biotechnol 2011, in press. [Google Scholar]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar]
- Barbolina, M.V.; Moss, N.M.; Westfall, S.D.; Liu, Y.; Burkhalter, R.J.; Marga, F.; Forgacs, G.; Hudson, L.G.; Stack, M.S. Microenvironmental regulation of ovarian cancer metastasis. Cancer Treat. Res 2009, 149, 319–334. [Google Scholar]
- Lathia, J.D.; Heddleston, J.M.; Venere, M.; Rich, J.N. Deadly teamwork: Neural cancer stem cells and the tumor microenvironment. Cell Stem Cell 2011, 8, 482–485. [Google Scholar]
- Jagannathan, N.R.; Bhujwalla, Z.M. Tumor microenvironment in cancer treatment and metastasis. NMR Biomed 2011, 24, 559–560. [Google Scholar]
- Cichon, M.A.; Degnim, A.C.; Visscher, D.W.; Radisky, D.C. Microenvironmental influences that drive progression from benign breast disease to invasive breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 389–397. [Google Scholar]
- Malchenko, S.; Galat, V.; Seftor, E.A.; Vanin, E.F.; Costa, F.F.; Seftor, R.E.; Soares, M.B.; Hendrix, M.J. Cancer hallmarks in induced pluripotent cells: New insights. J. Cell. Physiol 2010, 225, 390–393. [Google Scholar]
- Topczewska, J.M.; Postovit, L.M.; Margaryan, N.V.; Sam, A.; Hess, A.R.; Wheaton, W.W.; Nickoloff, B.J.; Topczewski, J.; Hendrix, M.J. Embryonic and tumorigenic pathways converge via nodal signaling: Role in melanoma aggressiveness. Nat. Med 2006, 12, 925–932. [Google Scholar]
- Abbott, D.E.; Postovit, L.M.; Seftor, E.A.; Margaryan, N.V.; Seftor, R.E.; Hendrix, M.J. Exploiting the convergence of embryonic and tumorigenic signaling pathways to develop new therapeutic targets. Stem Cell Rev 2007, 3, 68–78. [Google Scholar]
- Postovit, L.M.; Costa, F.F.; Bischof, J.M.; Seftor, E.A.; Wen, B.; Seftor, R.E.; Feinberg, A.P.; Soares, M.B.; Hendrix, M.J. The commonality of plasticity underlying multipotent tumor cells and embryonic stem cells. J. Cell. Biochem 2007, 101, 908–917. [Google Scholar]
- Ingber, D.E. Can cancer be reversed by engineering the tumor microenvironment? Semin. Cancer Biol 2008, 18, 356–364. [Google Scholar]
- Giuffrida, D.; Rogers, I.M.; Nagy, A.; Calogero, A.E.; Brown, T.J.; Casper, R.F. Human embryonic stem cells secrete soluble factors that inhibit cancer cell growth. Cell Prolif 2009, 42, 788–798. [Google Scholar]
- Costa, F.F.; Seftor, E.A.; Bischof, J.M.; Kirschmann, D.A.; Strizzi, L.; Arndt, K.; de Fatima Bonaldo, M.; Soares, M.B.; Hendrix, M.J. Epigenetically reprogramming metastatic tumor cells with an embryonic microenvironment. Epigenomics 2009, 1, 387–398. [Google Scholar]
- Postovit, L.M.; Margaryan, N.V.; Seftor, E.A.; Kirschmann, D.A.; Lipavsky, A.; Wheaton, W.W.; Abbott, D.E.; Seftor, R.E.; Hendrix, M.J. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4329–4334. [Google Scholar]
- Kulesa, P.M.; Kasemeier-Kulesa, J.C.; Teddy, J.M.; Margaryan, N.V.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc. Natl. Acad. Sci. USA 2006, 103, 3752–3757. [Google Scholar]
- Kim, M.O.; Kim, S.H.; Oi, N.; Lee, M.H.; Yu, D.H.; Kim, D.J.; Cho, E.J.; Bode, A.M.; Cho, Y.Y.; Bowden, T.G.; et al. Embryonic stem-cell-preconditioned microenvironment induces loss of cancer cell properties in human melanoma cells. Pigment Cell Melanoma Res 2011, 24, 922–931. [Google Scholar]
- Tzukerman, M.; Rosenberg, T.; Reiter, I.; Ben-Eliezer, S.; Denkberg, G.; Coleman, R.; Reiter, Y.; Skorecki, K. The influence of a human embryonic stem cell-derived microenvironment on targeting of human solid tumor xenografts. Cancer Res 2006, 66, 3792–3801. [Google Scholar]
- Wang, Z.; Zhang, L.; Yeung, T.K.; Chen, X. Endocytosis deficiency of epidermal growth factor (egf) receptor-erbb2 heterodimers in response to egf stimulation. Mol. Biol. Cell 1999, 10, 1621–1636. [Google Scholar]
- Caldon, C.E.; Sutherland, R.L.; Musgrove, E. Cell cycle proteins in epithelial cell differentiation: Implications for breast cancer. Cell Cycle 2010, 9, 1918–1928. [Google Scholar]
- Lee, E.Y.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol 2010, 2, a003236. [Google Scholar]
- Hollestelle, A.; Nagel, J.H.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat 2010, 121, 53–64. [Google Scholar]
- Mackay, A.; Tamber, N.; Fenwick, K.; Iravani, M.; Grigoriadis, A.; Dexter, T.; Lord, C.J.; Reis-Filho, J.S.; Ashworth, A. A high-resolution integrated analysis of genetic and expression profiles of breast cancer cell lines. Breast Cancer Res. Treat 2009, 118, 481–498. [Google Scholar]
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.L.; Girard, L.; Gandhi, J.; Kwei, K.A.; Hernandez-Boussard, T.; Wang, P.; Gazdar, A.F.; et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One 2009, 4, e6146:1–e6146:16. [Google Scholar]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar]
- Herrera-Gayol, A.; Jothy, S. Adhesion proteins in the biology of breast cancer: Contribution of cd44. Exp. Mol. Pathol 1999, 66, 149–156. [Google Scholar]
- Sethi, S.; Sarkar, F.H.; Ahmed, Q.; Bandyopadhyay, S.; Nahleh, Z.A.; Semaan, A.; Sakr, W.; Munkarah, A.; Ali-Fehmi, R. Molecular markers of epithelial-to-mesenchymal transition are associated with tumor aggressiveness in breast carcinoma. Transl. Oncol 2011, 4, 222–226. [Google Scholar]
- Taherian, A.; Li, X.; Liu, Y.; Haas, T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 2011, 11, 293:1–293:15. [Google Scholar]
- Creighton, C.J.; Chang, J.C.; Rosen, J.M. Epithelial-mesenchymal transition (emt) in tumor-initiating cells and its clinical implications in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 253–260. [Google Scholar]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; Yao, H.; et al. Ccl18 from tumor-associated macrophages promotes breast cancer metastasis via pitpnm3. Cancer Cell 2011, 19, 541–555. [Google Scholar]
- Li, J.Y.; Ou, Z.L.; Yu, S.J.; Gu, X.L.; Yang, C.; Chen, A.X.; Di, G.H.; Shen, Z.Z.; Shao, Z.M. The chemokine receptor ccr4 promotes tumor growth and lung metastasis in breast cancer. Breast Cancer Res. Treat 2011, in press. [Google Scholar]
- Johnson-Holiday, C.; Singh, R.; Johnson, E.; Singh, S.; Stockard, C.R.; Grizzle, W.E.; Lillard, J.W., Jr. Ccl25 mediates migration, invasion and matrix metalloproteinase expression by breast cancer cells in a ccr9-dependent fashion. Int. J. Oncol 2011, 38, 1279–1285. [Google Scholar]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar]
- Patsialou, A.; Wyckoff, J.; Wang, Y.; Goswami, S.; Stanley, E.R.; Condeelis, J.S. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res 2009, 69, 9498–9506. [Google Scholar]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res 2008, 68, 989–997. [Google Scholar]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: Interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res 2011, 13. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Tnf-alpha/nf-kappab/snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar]
- Weaver, A.M. Invadopodia: Specialized cell structures for cancer invasion. Clin. Exp. Metastasis 2006, 23, 97–105. [Google Scholar]
- Jiang, P.; Enomoto, A.; Takahashi, M. Cell biology of the movement of breast cancer cells: Intracellular signalling and the actin cytoskeleton. Cancer Lett 2009, 284, 122–130. [Google Scholar]
- Bae, Y.H.; Ding, Z.; Das, T.; Wells, A.; Gertler, F.; Roy, P. Profilin1 regulates pi(3,4)p2 and lamellipodin accumulation at the leading edge thus influencing motility of mda-mb-231 cells. Proc. Natl. Acad. Sci. USA 2010, 107, 21547–21552. [Google Scholar]
- Bae, Y.H.; Ding, Z.; Zou, L.; Wells, A.; Gertler, F.; Roy, P. Loss of profilin-1 expression enhances breast cancer cell motility by ena/vasp proteins. J. Cell. Physiol 2009, 219, 354–364. [Google Scholar]
- Gertler, F.; Condeelis, J. Metastasis: Tumor cells becoming menacing. Trends Cell Biol 2011, 21, 81–90. [Google Scholar]
- Yilmaz, M.; Christofori, G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res 2010, 8, 629–642. [Google Scholar]
- Sahai, E. Illuminating the metastatic process. Nat. Rev. Cancer 2007, 7, 737–749. [Google Scholar]
- Longatto Filho, A.; Lopes, J.M.; Schmitt, F.C. Angiogenesis and breast cancer. J. Oncol 2010, 2010, 576384:1–576384:7. [Google Scholar]
- Carpini, J.D.; Karam, A.K.; Montgomery, L. Vascular endothelial growth factor and its relationship to the prognosis and treatment of breast, ovarian, and cervical cancer. Angiogenesis 2010, 13, 43–58. [Google Scholar]
- Chakraborty, G.; Rangaswami, H.; Jain, S.; Kundu, G.C. Hypoxia regulates cross-talk between syk and lck leading to breast cancer progression and angiogenesis. J. Biol. Chem 2006, 281, 11322–11331. [Google Scholar]
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol 2008, 84, 623–630. [Google Scholar]
- Ronnov-Jessen, L.; Bissell, M.J. Breast cancer by proxy: Can the microenvironment be both the cause and consequence? Trends Mol. Med 2009, 15, 5–13. [Google Scholar]
- McSherry, E.A.; Donatello, S.; Hopkins, A.M.; McDonnell, S. Molecular basis of invasion in breast cancer. Cell. Mol. Life Sci 2007, 64, 3201–3218. [Google Scholar]
- Hu, M.; Polyak, K. Molecular characterisation of the tumour microenvironment in breast cancer. Eur. J. Cancer 2008, 44, 2760–2765. [Google Scholar]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212. [Google Scholar]
- Kohrmann, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Anacker, J. Expression of matrix metalloproteinases (mmps) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer 2009, 9, 188:1–188:20. [Google Scholar]
- Bostrom, P.; Soderstrom, M.; Vahlberg, T.; Soderstrom, K.O.; Roberts, P.J.; Carpen, O.; Hirsimaki, P. Mmp-1 expression has an independent prognostic value in breast cancer. BMC Cancer 2011, 11, 348:1–348:8. [Google Scholar]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of mmp-9 and hif-1alpha in breast cancer cells under hypoxic conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar]
- Zhang, B.; Liu, Y.X.; Cao, W.F.; Cao, X.C.; Ning, L.S.; Hao, X.S. Relationship between the expression of matrix metalloproteinase-13 protein and other biomarkers, prognosis in invasive breast cancer. Zhonghua Bing Li Xue Za Zhi 2008, 37, 471–476. [Google Scholar]
- Stark, A.M.; Anuszkiewicz, B.; Mentlein, R.; Yoneda, T.; Mehdorn, H.M.; Held-Feindt, J. Differential expression of matrix metalloproteinases in brain- and bone-seeking clones of metastatic mda-mb-231 breast cancer cells. J. Neurooncol 2007, 81, 39–48. [Google Scholar]
- Denhardt, D.T.; Feng, B.; Edwards, D.R.; Cocuzzi, E.T.; Malyankar, U.M. Tissue inhibitor of metalloproteinases (timp, aka epa): Structure, control of expression and biological functions. Pharmacol. Ther 1993, 59, 329–341. [Google Scholar]
- Nguyen, Q.; Willenbrock, F.; Cockett, M.I.; O’Shea, M.; Docherty, A.J.; Murphy, G. Different domain interactions are involved in the binding of tissue inhibitors of metalloproteinases to stromelysin-1 and gelatinase a. Biochemistry 1994, 33, 2089–2095. [Google Scholar]
- Vempati, P.; Karagiannis, E.D.; Popel, A.S. A biochemical model of matrix metalloproteinase 9 activation and inhibition. J. Biol. Chem 2007, 282, 37585–37596. [Google Scholar]
- O’Connell, J.P.; Willenbrock, F.; Docherty, A.J.; Eaton, D.; Murphy, G. Analysis of the role of the cooh-terminal domain in the activation, proteolytic activity, and tissue inhibitor of metalloproteinase interactions of gelatinase b. J. Biol. Chem 1994, 269, 14967–14973. [Google Scholar]
- Knauper, V.; Lopez-Otin, C.; Smith, B.; Knight, G.; Murphy, G. Biochemical characterization of human collagenase-3. J. Biol. Chem 1996, 271, 1544–1550. [Google Scholar]
- Stratmann, B.; Farr, M.; Tschesche, H. Characterization of c-terminally truncated human tissue inhibitor of metalloproteinases-4 expressed in pichia pastoris. Biol. Chem 2001, 382, 987–991. [Google Scholar]
- Hardy, K.M.; Booth, B.W.; Hendrix, M.J.; Salomon, D.S.; Strizzi, L. Erbb/egf signaling and emt in mammary development and breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 191–199. [Google Scholar]
- Raja, W.K.; Gligorijevic, B.; Wyckoff, J.; Condeelis, J.S.; Castracane, J. A new chemotaxis device for cell migration studies. Integr. Biol. (Camb) 2010, 2, 696–706. [Google Scholar]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J 2011, 437, 199–213. [Google Scholar]
- Scollen, S.; Luccarini, C.; Baynes, C.; Driver, K.; Humphreys, M.K.; Garcia-Closas, M.; Figueroa, J.; Lissowska, J.; Pharoah, P.D.; Easton, D.F.; et al. Tgf-beta signaling pathway and breast cancer susceptibility. Cancer Epidemiol. Biomark. Prev 2011, 20, 1112–1119. [Google Scholar]
- Swaminathan, V.; Mythreye, K.; O’Brien, E.T.; Berchuck, A.; Blobe, G.C.; Superfine, R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res 2011, 71, 5075–5080. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Wendt, M.K.; Taylor, M.A.; Schiemann, B.J.; Schiemann, W.P. Down-regulation of epithelial cadherin is required to initiate metastatic outgrowth of breast cancer. Mol. Biol. Cell 2011, 22, 2423–2435. [Google Scholar]
- Morozevich, G.; Kozlova, N.; Cheglakov, I.; Ushakova, N.; Berman, A. Integrin alpha5beta1 controls invasion of human breast carcinoma cells by direct and indirect modulation of mmp-2 collagenase activity. Cell Cycle 2009, 8, 2219–2225. [Google Scholar]
- Polyak, K.; Kalluri, R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb. Perspect. Biol 2010, 2, a003244:1–a003244:14. [Google Scholar]
- Robinson, B.D.; Sica, G.L.; Liu, Y.F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor microenvironment of metastasis in human breast carcinoma: A potential prognostic marker linked to hematogenous dissemination. Clin. Cancer Res 2009, 15, 2433–2441. [Google Scholar]
- Yizraeli, M.L.; Weihs, D. Time-dependent micromechanical responses of breast cancer cells and adjacent fibroblasts to electric treatment. Cell Biochem. Biophys 2011, in press. [Google Scholar]
- Kashani, I.; Barvarestani, M.; Etesam, F.; Shokrgozar, M.; Abdolvahabi, M.; Haddad, P.; Noori Mokohi, M.H.; Hosseinf, M. Human preadipocytes inhibit proliferation of MCF-7 breast cancer cell line. Acta Medica Iran 2006, 44, 291–298. [Google Scholar]
- Krause, S.; Maffini, M.V.; Soto, A.M.; Sonnenschein, C. The microenvironment determines the breast cancer cells’ phenotype: Organization of mcf7 cells in 3d cultures. BMC Cancer 2010, 10, 263:1–263:13. [Google Scholar]
- Krause, S.; Maffini, M.V.; Soto, A.M.; Sonnenschein, C. A novel 3d in vitro culture model to study stromal-epithelial interactions in the mammary gland. Tissue Eng. Part C Methods 2008, 14, 261–271. [Google Scholar]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res 2011, 71, 2455–2465. [Google Scholar]
- Poczobutt, J.M.; Tentler, J.; Lu, X.; Schedin, P.J.; Gutierrez-Hartmann, A. Benign mammary epithelial cells enhance the transformed phenotype of human breast cancer cells. BMC Cancer 2010, 10, 373:1–373:17. [Google Scholar]
- Sasser, A.K.; Mundy, B.L.; Smith, K.M.; Studebaker, A.W.; Axel, A.E.; Haidet, A.M.; Fernandez, S.A.; Hall, B.M. Human bone marrow stromal cells enhance breast cancer cell growth rates in a cell line-dependent manner when evaluated in 3d tumor environments. Cancer Lett 2007, 254, 255–264. [Google Scholar]
- Ingthorsson, S.; Sigurdsson, V.; Fridriksdottir, A., Jr.; Jonasson, J.G.; Kjartansson, J.; Magnusson, M.K.; Gudjonsson, T. Endothelial cells stimulate growth of normal and cancerous breast epithelial cells in 3d culture. BMC Res. Notes 2010, 3, 184:1–184:12. [Google Scholar]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell 2005, 7, 505–512. [Google Scholar]
- Yaar, M.; Eller, M.S.; Panova, I.; Kubera, J.; Wee, L.H.; Cowan, K.H.; Gilchrest, B.A. Telomeric DNA induces apoptosis and senescence of human breast carcinoma cells. Breast Cancer Res 2007, 9, R13:1–R13:13. [Google Scholar]
- Huang, M.; Whang, P.; Lewicki, P.; Mitchell, B.S. Cyclopentenyl cytosine induces senescence in breast cancer cells through the nucleolar stress response and activation of p53. Mol. Pharmacol 2011, 80, 40–48. [Google Scholar]
- Callahan, R.; Hurvitz, S. Human epidermal growth factor receptor-2-positive breast cancer: Current management of early, advanced, and recurrent disease. Curr. Opin. Obstet. Gynecol 2011, 23, 37–43. [Google Scholar]
- Sun, J. Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J. Signal Transduct 2010, 2010, 985132:1–985132:7. [Google Scholar]
- Koziczak, M.; Holbro, T.; Hynes, N.E. Blocking of fgfr signaling inhibits breast cancer cell proliferation through downregulation of d-type cyclins. Oncogene 2004, 23, 3501–3508. [Google Scholar]
- Eckert, M.A.; Yang, J. Targeting invadopodia to block breast cancer metastasis. Oncotarget 2011, 2, 562–568. [Google Scholar]
- Subbaram, S.; Dipersio, C.M. Integrin alpha3beta1 as a breast cancer target. Expert Opin. Ther. Targets 2011, in press. [Google Scholar]
- Ju, J.H.; Jang, K.; Lee, K.M.; Kim, M.; Kim, J.; Yi, J.Y.; Noh, D.Y.; Shin, I. Cd24 enhances DNA damage-induced apoptosis by modulating nf-{kappa}b signaling in cd44 expressing breast cancer cells. Carcinogenesis 2011. [Google Scholar] [CrossRef]
- Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The oct4 and nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet 2006, 38, 431–440. [Google Scholar]
- Pan, G.; Thomson, J.A. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res 2007, 17, 42–49. [Google Scholar]
- Hu, J.; Qin, K.; Zhang, Y.; Gong, J.; Li, N.; Lv, D.; Xiang, R.; Tan, X. Downregulation of transcription factor oct4 induces an epithelial-to-mesenchymal transition via enhancement of ca(2+) influx in breast cancer cells. Biochem. Biophys. Res. Commun 2011, 411, 786–791. [Google Scholar]
- Guo, Y.; Graham-Evans, B.; Broxmeyer, H.E. Murine embryonic stem cells secrete cytokines/growth modulators that enhance cell survival/anti-apoptosis and stimulate colony formation of murine hematopoietic progenitor cells. Stem Cells 2006, 24, 850–856. [Google Scholar]
- Llanes-Fernández, L.; Álvarez-Goyanes, R.I.; del Carmen Arango-, M.; Alcocer-González, J.M.; Mojarrieta, J.C.; Pérez, X.E.; López, M.O.; Odio, S.F.; Camacho-Rodríguez, R.; Guerra-Yi, M.E.; et al. Relationship between il-10 and tumor markers in breast cancer patients. Breast 2006, 15, 482–489. [Google Scholar]
- Gerger, A.; Renner, W.; Langsenlehner, T.; Hofmann, G.; Knechtel, G.; Szkandera, J.; Samonigg, H.; Krippl, P.; Langsenlehner, U. Association of interleukin-10 gene variation with breast cancer prognosis. Breast Cancer Res. Treat 2010, 119, 701–705. [Google Scholar]
- Lacroix, M.; Siwek, B.; Marie, P.J.; Body, J.J. Production and regulation of interleukin-11 by breast cancer cells. Cancer Lett 1998, 127, 29–35. [Google Scholar]
- Hanavadi, S.; Martin, T.; Watkins, G.; Mansel, R.; Jiang, W. Expression of interleukin 11 and its receptor and their prognostic value in human breast cancer. Ann. Surg. Oncol 2006, 13, 802–808. [Google Scholar]
- Singer, C.F.; Kronsteiner, N.; Hudelist, G.; Marton, E.; Walter, I.; Kubista, M.; Czerwenka, K.; Schreiber, M.; Seifert, M.; Kubista, E. Interleukin 1 system and sex steroid receptor expression in human breast cancer: Interleukin 1alpha protein secretion is correlated with malignant phenotype. Clin. Cancer Res 2003, 9, 4877–4883. [Google Scholar]
- Liu, J.; Spence, M.J.; Wallace, P.M.; Forcier, K.; Hellstrom, I.; Vestal, R.E. Oncostatin m-specific receptor mediates inhibition of breast cancer cell growth and down-regulation of the c-myc proto-oncogene. Cell Growth Differ 1997, 8, 667–676. [Google Scholar]
- Liu, J.; Hadjokas, N.; Mosley, B.; Estrov, Z.; Spence, M.J.; Vestal, R.E. Oncostatin m-specific receptor expression and function in regulating cell proliferation of normal and malignant mammary epithelial cells. Cytokine 1998, 10, 295–302. [Google Scholar]
- Jorcyk, C.L.; Holzer, R.G.; Ryan, R.E. Oncostatin m induces cell detachment and enhances the metastatic capacity of t-47d human breast carcinoma cells. Cytokine 2006, 33, 323–336. [Google Scholar]
- Ulivi, P.; Zoli, W.; Medri, L.; Amadori, D.; Saragoni, L.; Barbanti, F.; Calistri, D.; Silvestrini, R. C-kit and scf expression in normal and tumor breast tissue. Breast Cancer Res. Treat 2004, 83, 33–42. [Google Scholar]
- Hines, S.J.; Litz, J.S.; Krystal, G.W. Coexpression of c-kit and stem cell factor in breast cancer results in enhanced sensitivity to members of the egf family of growth factors. Breast Cancer Res. Treat 1999, 58, 1–10. [Google Scholar]
- Roland, C.L.; Dineen, S.P.; Lynn, K.D.; Sullivan, L.A.; Dellinger, M.T.; Sadegh, L.; Sullivan, J.P.; Shames, D.S.; Brekken, R.A. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol. Cancer Ther 2009, 8, 1761–1771. [Google Scholar]
- Bieche, I.; Chavey, C.; Andrieu, C.; Busson, M.; Vacher, S.; Le Corre, L.; Guinebretiere, J.M.; Burlinchon, S.; Lidereau, R.; Lazennec, G. Cxc chemokines located in the 4q21 region are up-regulated in breast cancer. Endocr. Relat. Cancer 2007, 14, 1039–1052. [Google Scholar]
- Walser, T.C.; Rifat, S.; Ma, X.; Kundu, N.; Ward, C.; Goloubeva, O.; Johnson, M.G.; Medina, J.C.; Collins, T.L.; Fulton, A.M. Antagonism of cxcr3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res 2006, 66, 7701–7707. [Google Scholar]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massague, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. Ccl2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar]
- Nath, A.; Chattopadhya, S.; Chattopadhyay, U.; Sharma, N.K. Macrophage inflammatory protein (mip)1alpha and mip1beta differentially regulate release of inflammatory cytokines and generation of tumoricidal monocytes in malignancy. Cancer Immunol. Immunother 2006, 55, 1534–1541. [Google Scholar]
- Ohara, M.; Yamaguchi, Y.; Matsuura, K.; Murakami, S.; Arihiro, K.; Okada, M. Possible involvement of regulatory t cells in tumor onset and progression in primary breast cancer. Cancer Immunol. Immunother 2009, 58, 441–447. [Google Scholar]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell. Biochem 2010, 111, 1138–1148. [Google Scholar]
- Gomes, E.M.; Rodrigues, M.S.; Phadke, A.P.; Butcher, L.D.; Starling, C.; Chen, S.; Chang, D.; Hernandez-Alcoceba, R.; Newman, J.T.; Stone, M.J.; et al. Antitumor activity of an oncolytic adenoviral-cd40 ligand (cd154) transgene construct in human breast cancer cells. Clin. Cancer Res 2009, 15, 1317–1325. [Google Scholar]
- Lee, S.J.; Yoo, H.J.; Bae, Y.S.; Kim, H.J.; Lee, S.T. Timp-1 inhibits apoptosis in breast carcinoma cells via a pathway involving pertussis toxin-sensitive g protein and c-src. Biochem. Biophys. Res. Commun 2003, 312, 1196–1201. [Google Scholar]
- Michalska, A.E. Chapter 1, Unit1C.3 Isolation and propagation of mouse embryonic fibroblasts and preparation of mouse embryonic feeder layer cells. Current Protocols in Stem Cell Biology 2007. [Google Scholar]
- Amit, M.; Margulets, V.; Segev, H.; Shariki, K.; Laevsky, I.; Coleman, R.; Itskovitz-Eldor, J. Human feeder layers for human embryonic stem cells. Biol. Reprod 2003, 68, 2150–2156. [Google Scholar]
- Lin, S.; Talbot, P. Methods for culturing mouse and human embryonic stem cells. Methods Mol. Biol 2011, 690, 31–56. [Google Scholar]
- Abraham, S.; Sheridan, S.D.; Miller, B.; Rao, R.R. Stable propagation of human embryonic and induced pluripotent stem cells on decellularized human substrates. Biotechnol. Prog 2010, 26, 1126–1134. [Google Scholar]
- Hirai, H.; Karian, P.; Kikyo, N. Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem. J 2011, 438, 11–23. [Google Scholar]
- Levenstein, M.E.; Ludwig, T.E.; Xu, R.H.; Llanas, R.A.; Van Den Heuvel-Kramer, K.; Manning, D.; Thomson, J.A. Basic fibroblast growth factor support of human embryonic stem cell self-renewal. Stem Cells 2006, 24, 568–574. [Google Scholar]
- Wang, L.; Schulz, T.C.; Sherrer, E.S.; Dauphin, D.S.; Shin, S.; Nelson, A.M.; Ware, C.B.; Zhan, M.; Song, C.Z.; Chen, X.; et al. Self-renewal of human embryonic stem cells requires insulin-like growth factor-1 receptor and erbb2 receptor signaling. Blood 2007, 110, 4111–4119. [Google Scholar]
- Gerecht, S.; Burdick, J.A.; Ferreira, L.S.; Townsend, S.A.; Langer, R.; Vunjak-Novakovic, G. Hyaluronic acid hydrogel for controlled self-renewal and differentiation of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 11298–11303. [Google Scholar]
- Siti-Ismail, N.; Bishop, A.E.; Polak, J.M.; Mantalaris, A. The benefit of human embryonic stem cell encapsulation for prolonged feeder-free maintenance. Biomaterials 2008, 29, 3946–3952. [Google Scholar]
- Wang, X.; Wang, W.; Ma, J.; Guo, X.; Yu, X.; Ma, X. Proliferation and differentiation of mouse embryonic stem cells in apa microcapsule: A model for studying the interaction between stem cells and their niche. Biotechnol. Prog 2006, 22, 791–800. [Google Scholar]
- Li, Z.; Leung, M.; Hopper, R.; Ellenbogen, R.; Zhang, M. Feeder-free self-renewal of human embryonic stem cells in 3d porous natural polymer scaffolds. Biomaterials 2010, 31, 404–412. [Google Scholar]
- Raof, N.A.; Padgen, M.R.; Gracias, A.R.; Bergkvist, M.; Xie, Y. One-dimensional self-assembly of mouse embryonic stem cells using an array of hydrogel microstrands. Biomaterials 2011, 32, 4498–4505. [Google Scholar]
- Peng, S.; Hua, J.; Cao, X.; Wang, H. Gelatin induces trophectoderm differentiation of mouse embryonic stem cells. Cell Biol. Int 2011, 35, 587–591. [Google Scholar]
- Farzaneh, Z.; Pournasr, B.; Ebrahimi, M.; Aghdami, N.; Baharvand, H. Enhanced functions of human embryonic stem cell-derived hepatocyte-like cells on three-dimensional nanofibrillar surfaces. Stem Cell Rev 2010, 6, 601–610. [Google Scholar]
- Mahairaki, V.; Lim, S.H.; Christopherson, G.T.; Xu, L.; Nasonkin, I.; Yu, C.; Mao, H.Q.; Koliatsos, V.E. Nanofiber matrices promote the neuronal differentiation of human embryonic stem cell-derived neural precursors in vitro. Tissue Eng. Part A 2011, 17, 855–863. [Google Scholar]
- Smith, L.A.; Liu, X.; Hu, J.; Ma, P.X. The enhancement of human embryonic stem cell osteogenic differentiation with nano-fibrous scaffolding. Biomaterials 2010, 31, 5526–5535. [Google Scholar]
- Smith, L.A.; Liu, X.; Hu, J.; Ma, P.X. The influence of three-dimensional nanofibrous scaffolds on the osteogenic differentiation of embryonic stem cells. Biomaterials 2009, 30, 2516–2522. [Google Scholar]
- Kang, X.; Xie, Y.; Powell, H.M.; James Lee, L.; Belury, M.A.; Lannutti, J.J.; Kniss, D.A. Adipogenesis of murine embryonic stem cells in a three-dimensional culture system using electrospun polymer scaffolds. Biomaterials 2007, 28, 450–458. [Google Scholar]
- Xie, J.; Willerth, S.M.; Li, X.; Macewan, M.R.; Rader, A.; Sakiyama-Elbert, S.E.; Xia, Y. The differentiation of embryonic stem cells seeded on electrospun nanofibers into neural lineages. Biomaterials 2009, 30, 354–362. [Google Scholar]
- Schiele, N.R.; Corr, D.T.; Huang, Y.; Raof, N.A.; Xie, Y.; Chrisey, D.B. Laser-based direct-write techniques for cell printing. Biofabrication 2010, 2. [Google Scholar] [CrossRef]
- Postovit, L.M.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Influence of the microenvironment on melanoma cell fate determination and phenotype. Cancer Res 2006, 66, 7833–7836. [Google Scholar]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Kasemeier-Kulesa, J.; Kulesa, P.M.; Postovit, L.M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat. Rev. Cancer 2007, 7, 246–255. [Google Scholar]
- Kasemeier-Kulesa, J.C.; Teddy, J.M.; Postovit, L.M.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J.; Kulesa, P.M. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev. Dyn 2008, 237, 2657–2666. [Google Scholar]
- Illmensee, K.; Mintz, B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc. Natl. Acad. Sci. USA 1976, 73, 549–553. [Google Scholar]
- Pierce, G.B.; Pantazis, C.G.; Caldwell, J.E.; Wells, R.S. Specificity of the control of tumor formation by the blastocyst. Cancer Res 1982, 42, 1082–1087. [Google Scholar]
- Lee, L.M.; Seftor, E.A.; Bonde, G.; Cornell, R.A.; Hendrix, M.J. The fate of human malignant melanoma cells transplanted into zebrafish embryos: Assessment of migration and cell division in the absence of tumor formation. Dev. Dyn 2005, 233, 1560–1570. [Google Scholar]
- Diez-Torre, A.; Andrade, R.; Eguizabal, C.; Lopez, E.; Arluzea, J.; Silio, M.; Arechaga, J. Reprogramming of melanoma cells by embryonic microenvironments. Int. J. Dev. Biol 2009, 53, 1563–1568. [Google Scholar]
- Ma, F.; Zhou, L.; Fang, L.Q.; Bai, J.; Zhao, J.; Wang, Y.; Wang, Z.B. Effect of mid-late mouse fetus’ microenvironment on the growth of tumor cells after intrauterine transplantation. Cell Biol. Int 2007, 31, 592–598. [Google Scholar]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.; et al. Braf mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr. Biol 2005, 15, 249–254. [Google Scholar]
- Kelleher, F.; Fennelly, D.; Rafferty, M. Common critical pathways in embryogenesis and cancer. Acta Oncol 2006, 45, 375–388. [Google Scholar]
- Dreesen, O.; Brivanlou, A. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. Rep 2007, 3, 7–17. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Marcato, P.; Dean, C.A.; Pan, D.; Araslanova, R.; Gillis, M.; Joshi, M.; Helyer, L.; Pan, L.; Leidal, A.; Gujar, S.; et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform aldh1a3 and its expression is predictive of metastasis. Stem Cells 2011, 29, 32–45. [Google Scholar]
- Charafe-Jauffret, E.; Ginestier, C.; Birnbaum, D. Breast cancer stem cells: Tools and models to rely on. BMC Cancer 2009, 9, 202:1–202:10. [Google Scholar]
- Liu, S.; Ginestier, C.; Ou, S.J.; Clouthier, S.G.; Patel, S.H.; Monville, F.; Korkaya, H.; Heath, A.; Dutcher, J.; Kleer, C.G.; et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res 2011, 71, 614–624. [Google Scholar]
- Ginestier, C.; Charafe-Jauffret, E.; Birnbaum, D. Targeting breast cancer stem cells: Fishing season open! Breast Cancer Res 2010, 12, 312:–312:2. [Google Scholar]
- Liu, S.; Wicha, M.S. Targeting breast cancer stem cells. J. Clin. Oncol 2010, 28, 4006–4012. [Google Scholar]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest 2011, 121, 3804–3809. [Google Scholar]
- Velasco-Velazquez, M.A.; Popov, V.M.; Lisanti, M.P.; Pestell, R.G. The role of breast cancer stem cells in metastasis and therapeutic implications. Am. J. Pathol 2011, 179, 2–11. [Google Scholar]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008, 2, 333–344. [Google Scholar]
- Somervaille, T.C.P.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of mll myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar]
- Postovit, L.M.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. A three-dimensional model to study the epigenetic effects induced by the microenvironment of human embryonic stem cells. Stem Cells 2006, 24, 501–505. [Google Scholar]
- Abbott, D.E.; Bailey, C.M.; Postovit, L.M.; Seftor, E.A.; Margaryan, N.; Seftor, R.E.; Hendrix, M.J. The epigenetic influence of tumor and embryonic microenvironments: How different are they? Cancer Microenviron 2008, 1, 13–21. [Google Scholar]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet 2008, 40, 499–507. [Google Scholar]
- Raof, N.A.; Raja, W.K.; Castracane, J.; Xie, Y. Bioengineering embryonic stem cell microenvironments for exploring inhibitory effects on metastatic breast cancer cells. Biomaterials 2011, 32, 4130–4139. [Google Scholar]
- Kievit, F.M.; Florczyk, S.J.; Leung, M.C.; Veiseh, O.; Park, J.O.; Disis, M.L.; Zhang, M. Chitosan-alginate 3d scaffolds as a mimic of the glioma tumor microenvironment. Biomaterials 2010, 31, 5903–5910. [Google Scholar]
- Chen, M.C.; Gupta, M.; Cheung, K.C. Alginate-based microfluidic system for tumor spheroid formation and anticancer agent screening. Biomed. Microdevices 2010, 12, 647–654. [Google Scholar]
- Akeda, K.; Nishimura, A.; Satonaka, H.; Shintani, K.; Kusuzaki, K.; Matsumine, A.; Kasai, Y.; Masuda, K.; Uchida, A. Three-dimensional alginate spheroid culture system of murine osteosarcoma. Oncol. Rep 2009, 22, 997–1003. [Google Scholar]
- Plunkett, M.L.; Hailey, J.A. An in vivo quantitative angiogenesis model using tumor cells entrapped in alginate. Lab. Invest 1990, 62, 510–517. [Google Scholar]
- Yamada, K.M.; Cukierman, E. Modeling tissue morphogenesis and cancer in 3d. Cell 2007, 130, 601–610. [Google Scholar]
- Gautier, A.; Carpentier, B.; Dufresne, M.; Vu Dinh, Q.; Paullier, P.; Legallais, C. Impact of alginate type and bead diameter on mass transfers and the metabolic activities of encapsulated c3a cells in bioartificial liver applications. Eur. Cell Mater 2011, 21, 94–106. [Google Scholar]
- Strand, B.L.; Morch, Y.A.; Syvertsen, K.R.; Espevik, T.; Skjak-Braek, G. Microcapsules made by enzymatically tailored alginate. J. Biomed. Mater. Res. A 2003, 64, 540–550. [Google Scholar]
- Tan, C.S.; Jejurikar, A.; Rai, B.; Bostrom, T.; Lawrie, G.; Grondahl, L. Encapsulation of a glycosaminoglycan in hydroxyapatite/alginate capsules. J. Biomed. Mater. Res. A 2009, 91, 866–877. [Google Scholar]
- Hannouche, D.; Terai, H.; Fuchs, J.R.; Terada, S.; Zand, S.; Nasseri, B.A.; Petite, H.; Sedel, L.; Vacanti, J.P. Engineering of implantable cartilaginous structures from bone marrow-derived mesenchymal stem cells. Tissue Eng 2007, 13, 87–99. [Google Scholar]
- Wang, N.; Adams, G.; Buttery, L.; Falcone, F.H.; Stolnik, S. Alginate encapsulation technology supports embryonic stem cells differentiation into insulin-producing cells. J. Biotechnol 2009, 144, 304–312. [Google Scholar]
- Hwang, Y.S.; Cho, J.; Tay, F.; Heng, J.Y.; Ho, R.; Kazarian, S.G.; Williams, D.R.; Boccaccini, A.R.; Polak, J.M.; Mantalaris, A. The use of murine embryonic stem cells, alginate encapsulation, and rotary microgravity bioreactor in bone tissue engineering. Biomaterials 2009, 30, 499–507. [Google Scholar]
- Maguire, T.; Davidovich, A.E.; Wallenstein, E.J.; Novik, E.; Sharma, N.; Pedersen, H.; Androulakis, I.P.; Schloss, R.; Yarmush, M. Control of hepatic differentiation via cellular aggregation in an alginate microenvironment. Biotechnol. Bioeng 2007, 98, 631–644. [Google Scholar]
- Zhao, L.; Tang, M.; Weir, M.D.; Detamore, M.S.; Xu, H.H. Osteogenic media and rhbmp-2-induced differentiation of umbilical cord mesenchymal stem cells encapsulated in alginate microbeads and integrated in an injectable calcium phosphate-chitosan fibrous scaffold. Tissue Eng. Part A 2011, 17, 969–979. [Google Scholar]
- Bai, H.Y.; Chen, G.A.; Mao, G.H.; Song, T.R.; Wang, Y.X. Three step derivation of cartilage like tissue from human embryonic stem cells by 2d-3d sequential culture in vitro and further implantation in vivo on alginate/plga scaffolds. J. Biomed. Mater. Res. A 2010, 94, 539–546. [Google Scholar]
- Trivedi, N.; Keegan, M.; Steil, G.M.; Hollister-Lock, J.; Hasenkamp, W.M.; Colton, C.K.; Bonner-Weir, S.; Weir, G.C. Islets in alginate macrobeads reverse diabetes despite minimal acute insulin secretory responses. Transplantation 2001, 71, 203–211. [Google Scholar]
- Dufrane, D.; Goebbels, R.M.; Gianello, P. Alginate macroencapsulation of pig islets allows correction of streptozotocin-induced diabetes in primates up to 6 months without immunosuppression. Transplantation 2010, 90, 1054–1062. [Google Scholar]
- Dufrane, D.; Goebbels, R.M.; Saliez, A.; Guiot, Y.; Gianello, P. Six-month survival of microencapsulated pig islets and alginate biocompatibility in primates: Proof of concept. Transplantation 2006, 81, 1345–1353. [Google Scholar]
- Fritschy, W.M.; Wolters, G.H.; van Schilfgaarde, R. Effect of alginate-polylysine-alginate microencapsulation on in vitro insulin release from rat pancreatic islets. Diabetes 1991, 40, 37–43. [Google Scholar]
- Xu, M.; Wang, X.; Yan, Y.; Yao, R.; Ge, Y. An cell-assembly derived physiological 3d model of the metabolic syndrome, based on adipose-derived stromal cells and a gelatin/alginate/fibrinogen matrix. Biomaterials 2010, 31, 3868–3877. [Google Scholar]
- Ruvinov, E.; Leor, J.; Cohen, S. The promotion of myocardial repair by the sequential delivery of igf-1 and hgf from an injectable alginate biomaterial in a model of acute myocardial infarction. Biomaterials 2011, 32, 565–578. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Molecular Weight (kDa) | C (pg/mL) Secreted by ES Cells [104] | C (pg/mL) Released by Breast Cancer Cells | Effects on Breast Cancer Cells |
|---|---|---|---|---|
| CYTOKINES | 20–45 | |||
| IL-10 | 20 | +++ | ++ | Expressed in tumor samples [105] and associated with reduced disease-free survival [106] |
| IL-11 | 23 | ++ | +++ | Produced by breast cancer cells [107] and linked to poor survival [108] |
| IL-1α | 33 | ++ | +++ | Expressed in poorly differentiated, ERα-negative tumors [109] |
| M-CSF | 18.5 | +++ | ++++ | CSF-1/CSF-1R autocrine signaling contributed to the invasion phenotype of breast cancer [42] |
| OSM (Oncostatin M) | 28 | ++ | ++++++ | 0.1–100 ng/mL OSM: inhibited proliferation/changed cell morphology [110,111]; 20–50 ng/mL OSM: increased invasive potential [112] |
| SCF (Stem Cell Factor) | 45 | ++ | — | High expression of SCF and SCF-R in normal mammary samples and low in invasive tumors [113]; enhanced activation of the MAPK and PI3K pathways [114] |
| VEGF | 42 | +++ | +++ | Angiogenic effect [115] |
| CHEMOKINES | <13 | |||
| GCP-2/CXCL6 | 8 | ++ | — | Upregulated in breast cancer cells [116] |
| IP-10/CXCL10 | 10 | +++ | +++ | Promote metastasis in a murine model [117] |
| KC/GROα/CXCL 1 | 11.3 | +++ | — | Angiogenic effect [118] |
| MCP-1/CCL2 | 11–13 | +++ | +++ | Highly expressed in breast tumor [119] |
| MCP-3/CCL7 | 11 | ++ | — | Overexpressed in breast carcinoma patients [120] |
| MDC/CCL22 | 8.1 | ++ | — | Involved in breast cancer lung metastasis [121] |
| MIP-1β/CCL4 | 7.8 | ++ | +++ | Downregulated in breast carcinoma patients [120] |
| MIP-2/CXCL2 | 6 | + | +++ | Highly expressed in bone metastatic breast cancer [122] |
| OTHERS | >29 | |||
| CD 40 | 43 | ++ | — | Anti-tumor activity in breast cancer cells [123] |
| MMP-9 | 90 | ++++ | +++++ | Overexpressed in breast cancer cells [64] |
| TIMP-1 | 29 | +++++ | ++++++ | Inhibits breast cancer cell apoptosis [124] |
| Embryonic Microenvironments | Cancer Cells | Effects | References |
|---|---|---|---|
| Zebrafish embryo model | Human metastatic melanoma cells | Support cell survival and division with no tumor formation. | [150] |
| Embryonic chick model | Human metastatic melanoma cells | Revert the metastatic phenotype to its cell type of origin. | [24] |
| hESC-conditioned Matrigel | Human metastatic melanoma cells | Induce a melanocyte-like phenotype and significantly inhibit the in vitro invasiveness of cancer cells. | [166] |
| hESC-conditioned Matrigel | Human metastatic melanoma and breast cancer cells | Decrease Nodal expression and inhibit tumorigenesis. | [23] |
| hESC-conditioned Matrigel | Human metastatic melanoma cells | Decrease VE-Cadherin expression. | [167] |
| hESC-conditioned Matrigel | Human metastatic melanoma cells | Identify miRNAs up- and down- regulated in reprogramming of melanoma cells. | [22] |
| mESC-conditioned Matrigel | Human metastatic melanoma and breast cancer cells | Inhibit cell proliferation, decrease anchorage independence and induce senescence. | [25] |
| hESC-conditioned medium | Human epithelial ovarian, prostate, and breast cancer cells | Inhibit cell proliferation and cell cycle (increased cells in G1 and deceased cells in S and G2/M phase). | [21] |
| In vitro mouse embryo model | Human melanoma cells | Support the melanoma cell migration inside the embryo model in a way reminiscent of neural crest cells with no tumor growth. | [151] |
| Bioengineered mESC microenvironment | Rat metastatic breast cancer cells | Inhibit the growth and migration of breast cancer cells. | [169] |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raof, N.A.; Mooney, B.M.; Xie, Y. Bioengineering Embryonic Stem Cell Microenvironments for the Study of Breast Cancer. Int. J. Mol. Sci. 2011, 12, 7662-7691. https://doi.org/10.3390/ijms12117662
Raof NA, Mooney BM, Xie Y. Bioengineering Embryonic Stem Cell Microenvironments for the Study of Breast Cancer. International Journal of Molecular Sciences. 2011; 12(11):7662-7691. https://doi.org/10.3390/ijms12117662
Chicago/Turabian StyleRaof, Nurazhani Abdul, Bridget M. Mooney, and Yubing Xie. 2011. "Bioengineering Embryonic Stem Cell Microenvironments for the Study of Breast Cancer" International Journal of Molecular Sciences 12, no. 11: 7662-7691. https://doi.org/10.3390/ijms12117662
APA StyleRaof, N. A., Mooney, B. M., & Xie, Y. (2011). Bioengineering Embryonic Stem Cell Microenvironments for the Study of Breast Cancer. International Journal of Molecular Sciences, 12(11), 7662-7691. https://doi.org/10.3390/ijms12117662