Systematic Approaches towards the Development of Host-Directed Antiviral Therapeutics

Abstract
:1. Introduction
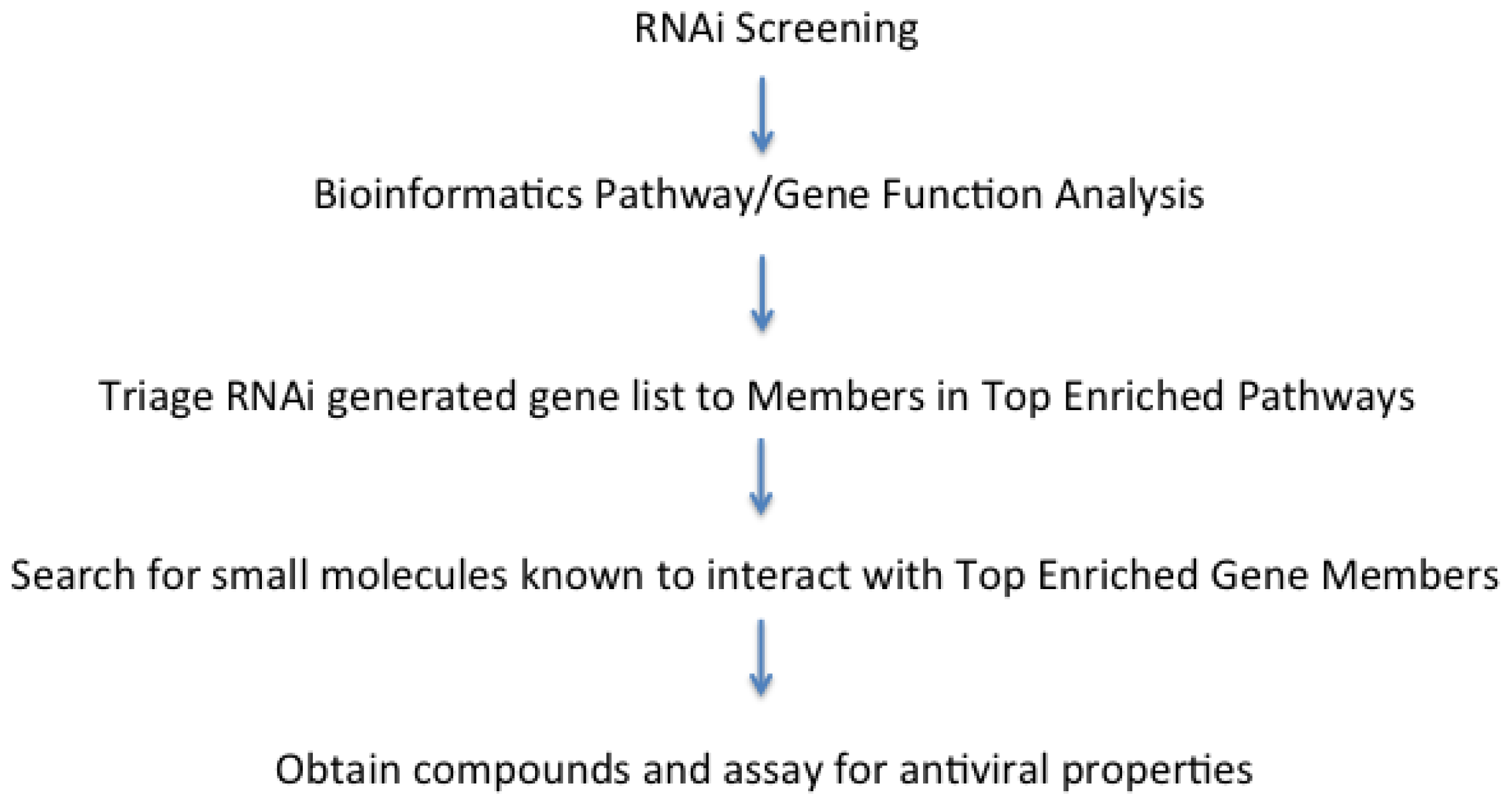
2. Methods to Analyze and Process Viral Host Factors Identified as Hits
3. Bioinformatics Approaches for Identifying Host-Factors Required for HIV Replication
3.1. Bioinformatics Approaches to Identify Host-Factors Required for HIV Virus Replication
3.2. Bioinformatics Approaches to Identify Host-Factors Required for Influenza Virus Replication
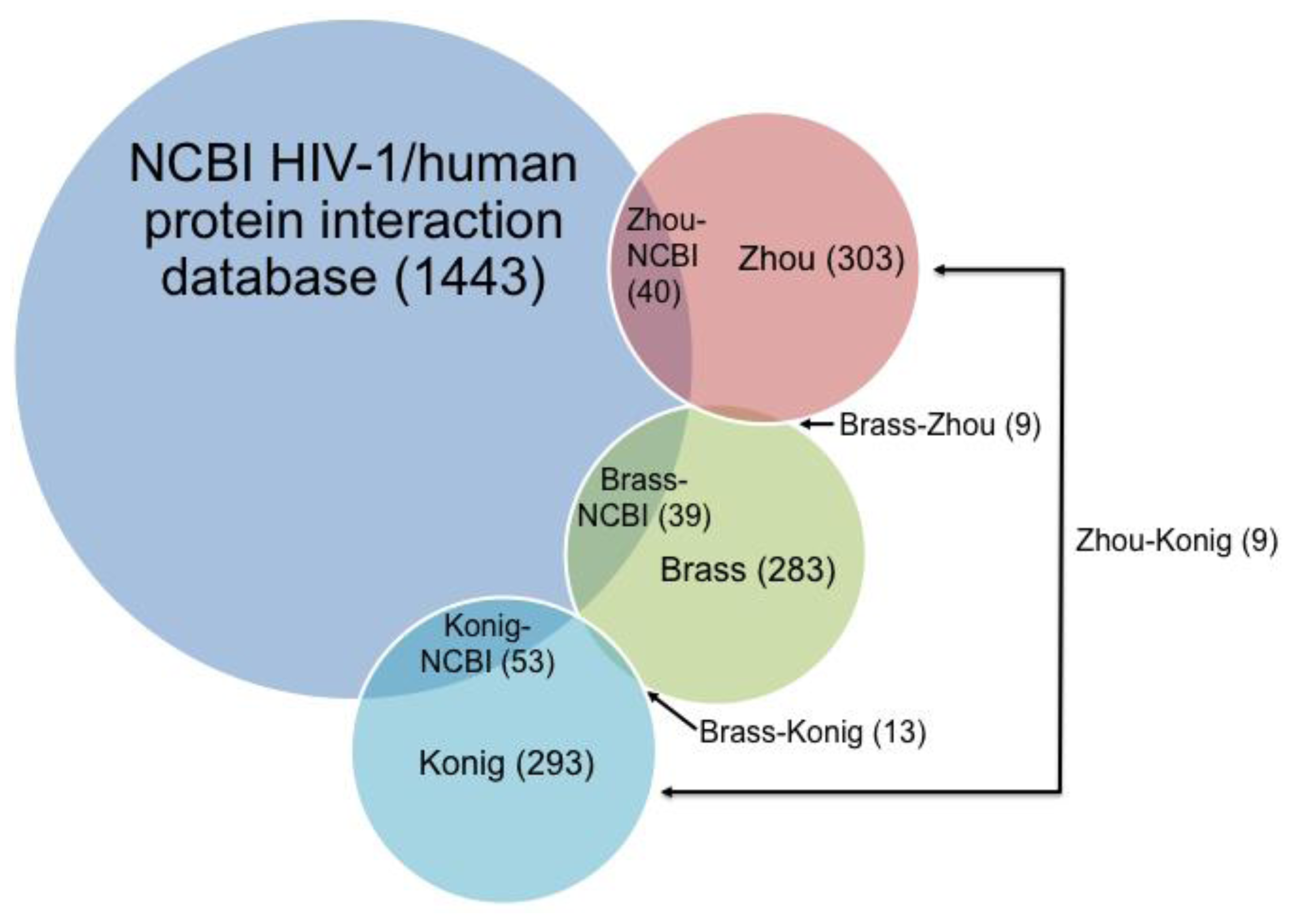
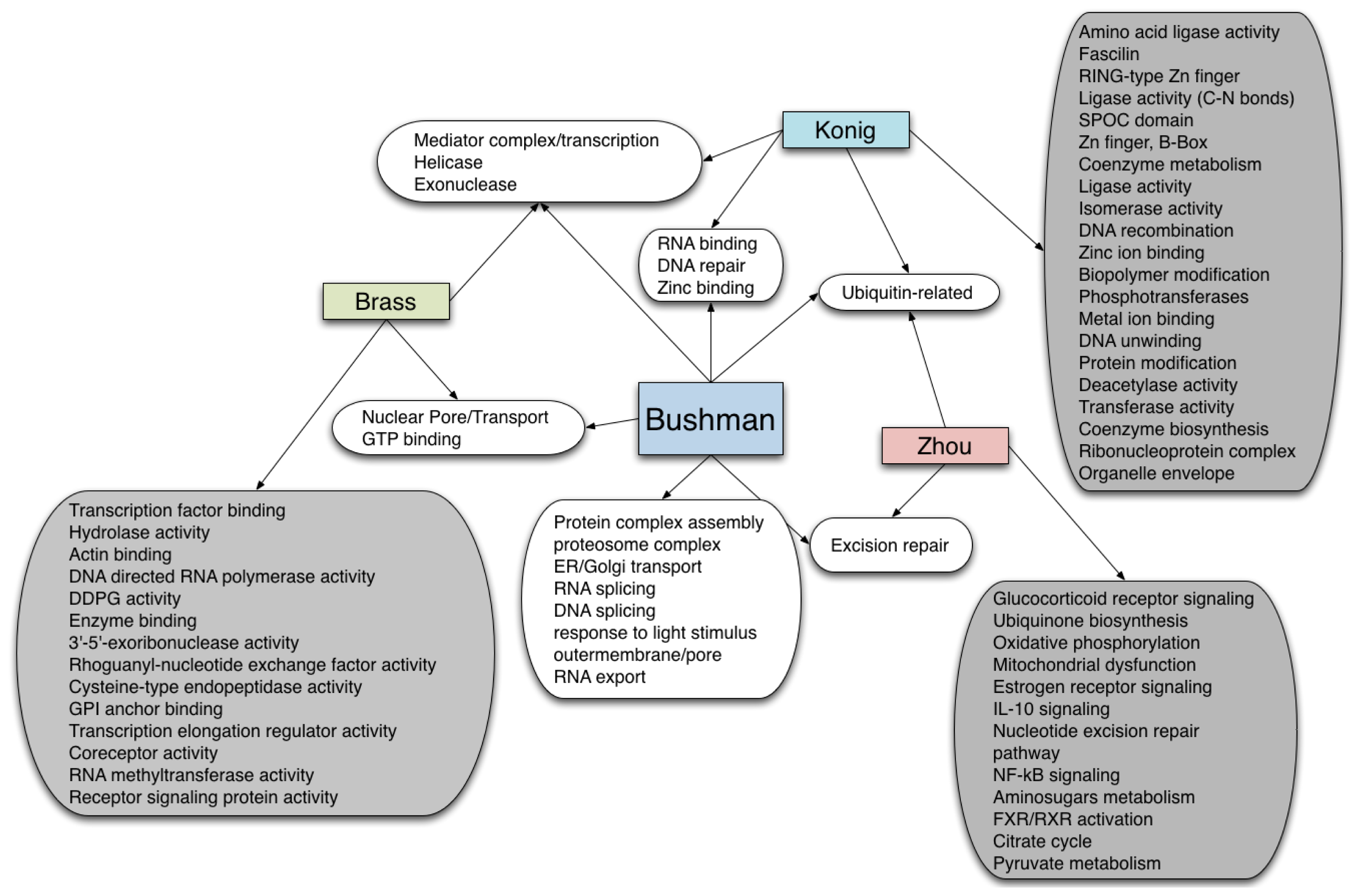
4. Pathway Database Comparisons: Same Source, Different Interpretation
5. Conclusions
Supporting Information

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
References
- Coen, DM; Richman, DD. Knipe, DM, Howley, PM, Eds.; Antiviral Agents. In Fields Virology, 5th ed; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 1, pp. 447–485. [Google Scholar]
- Moore, C; Galiano, M; Lackenby, A; Abdelrahman, T; Barnes, R; Evans, MR; Fegan, C; Froude, S; Hastings, M; Knapper, S; et al. Evidence of person-to-person transmission of oseltamivir-resistant pandemic influenza A(H1N1) 2009 virus in a hematology unit. J. Infect. Dis 2011, 203, 18–24. [Google Scholar]
- Sheu, TG; Fry, AM; Garten, RJ; Deyde, VM; Shwe, T; Bullion, L; Peebles, PJ; Li, Y; Klimov, AI; Gubareva, LV. Dual resistance to adamantanes and oseltamivir among seasonal influenza A(H1N1) viruses: 2008–2010. J. Infect. Dis 2011, 203, 13–17. [Google Scholar]
- Hayden, FG; de Jong, MD. Emerging influenza antiviral resistance threats. J. Infect. Dis 2011, 203, 6–10. [Google Scholar]
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781. [Google Scholar]
- Cegelski, L; Marshall, GR; Eldridge, GR; Hultgren, SJ. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol 2008, 6, 17–27. [Google Scholar]
- Schinazi, RF; Peters, J; Williams, CC; Chance, D; Nahmias, AJ. Effect of combinations of acyclovir with vidarabine or its 5′-monophosphate on herpes simplex viruses in cell culture and in mice. Antimicrob. Agents Chemother 1982, 22, 499–507. [Google Scholar]
- Kellam, P. Attacking pathogens through their hosts. Genome Biol 2006, 7, 201. [Google Scholar]
- Schwegmann, A; Brombacher, F. Host-directed drug targeting of factors hijacked by pathogens. Sci Signal 2008, 1, re8. [Google Scholar]
- Tan, SL; Ganji, G; Paeper, B; Proll, S; Katze, MG. Systems biology and the host response to viral infection. Nat. Biotechnol 2007, 25, 1383–1389. [Google Scholar]
- Koon, HB; Bubley, GJ; Pantanowitz, L; Masiello, D; Smith, B; Crosby, K; Proper, J; Weeden, W; Miller, TE; Chatis, P; et al. Imatinib-induced regression of AIDS-related Kaposi’s sarcoma. J. Clin. Oncol 2005, 23, 982–989. [Google Scholar]
- Salerno, D; Hasham, MG; Marshall, R; Garriga, J; Tsygankov, AY; Grana, X. Direct inhibition of CDK9 blocks HIV-1 replication without preventing T-cell activation in primary human peripheral blood lymphocytes. Gene 2007, 405, 65–78. [Google Scholar]
- Reeves, PM; Bommarius, B; Lebeis, S; McNulty, S; Christensen, J; Swimm, A; Chahroudi, A; Chavan, R; Feinberg, MB; Veach, D; et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat. Med 2005, 11, 731–739. [Google Scholar]
- Hirsch, AJ; Medigeshi, GR; Meyers, HL; DeFilippis, V; Fruh, K; Briese, T; Lipkin, WI; Nelson, JA. The Src family kinase c-Yes is required for maturation of West Nile virus particles. J. Virol 2005, 79, 11943–11951. [Google Scholar]
- Pearson, G; Robinson, F; Beers Gibson, T; Xu, BE; Karandikar, M; Berman, K; Cobb, MH. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev 2001, 22, 153–183. [Google Scholar]
- Pleschka, S; Wolff, T; Ehrhardt, C; Hobom, G; Planz, O; Rapp, UR; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol 2001, 3, 301–305. [Google Scholar]
- Zhu, H; Cong, JP; Yu, D; Bresnahan, WA; Shenk, TE. Inhibition of cyclooxygenase 2 blocks human cytomegalovirus replication. Proc. Natl. Acad. Sci. USA 2002, 99, 3932–3937. [Google Scholar]
- Goldman, JM; Druker, BJ. Chronic myeloid leukemia: current treatment options. Blood 2001, 98, 2039–2042. [Google Scholar]
- Ludwig, S. Targeting cell signalling pathways to fight the flu: Towards a paradigm change in anti-influenza therapy. J. Antimicrob. Chemother 2009, 64, 1–4. [Google Scholar]
- Newsome, TP; Scaplehorn, N; Way, M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science 2004, 306, 124–129. [Google Scholar]
- Kujime, K; Hashimoto, S; Gon, Y; Shimizu, K; Horie, T. p38 mitogen-activated protein kinase and c-jun-NH2-terminal kinase regulate RANTES production by influenza virus-infected human bronchial epithelial cells. J. Immunol 2000, 164, 3222–3228. [Google Scholar]
- Yoon, JJ; Chawla, D; Paal, T; Ndungu, M; Du, Y; Kurtkaya, S; Sun, A; Snyder, JP; Plemper, RK. High-throughput screening-based identification of paramyxovirus inhibitors. J. Biomol. Screen 2008, 13, 591–608. [Google Scholar]
- Bushell, KM; Sollner, C; Schuster-Boeckler, B; Bateman, A; Wright, GJ. Large-scale screening for novel low-affinity extracellular protein interactions. Genome Res 2008, 18, 622–630. [Google Scholar]
- Fields, S; Sternglanz, R. The two-hybrid system: an assay for protein-protein interactions. Trends Genet 1994, 10, 286–292. [Google Scholar]
- Chien, CT; Bartel, PL; Sternglanz, R; Fields, S. The two-hybrid system: A method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. USA 1991, 88, 9578–9582. [Google Scholar]
- Kaur, G; Roy, I. Therapeutic applications of aptamers. Expert Opin. Invest. Drugs 2008, 17, 43–60. [Google Scholar]
- Borghouts, C; Kunz, C; Groner, B. Peptide aptamer libraries. Comb. Chem. High Throughput Screen 2008, 11, 135–145. [Google Scholar]
- Grimm, D; Kay, MA. Therapeutic application of RNAi: Is mRNA targeting finally ready for prime time? J. Clin. Invest 2007, 117, 3633–3641. [Google Scholar]
- Fewell, GD; Schmitt, K. Vector-based RNAi approaches for stable, inducible and genome-wide screens. Drug Discov. Today 2006, 11, 975–982. [Google Scholar]
- Filipowicz, W; Bhattacharyya, SN; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet 2008, 9, 102–114. [Google Scholar]
- Stenvang, J; Kauppinen, S. MicroRNAs as targets for antisense-based therapeutics. Expert Opin. Biol. Ther 2008, 8, 59–81. [Google Scholar]
- Nguyen, DG; Yin, H; Zhou, Y; Wolff, KC; Kuhen, KL; Caldwell, JS. Identification of novel therapeutic targets for HIV infection through functional genomic cDNA screening. Virology 2007, 362, 16–25. [Google Scholar]
- Basha, S; Rai, P; Poon, V; Saraph, A; Gujraty, K; Go, MY; Sadacharan, S; Frost, M; Mogridge, J; Kane, RS. Polyvalent inhibitors of anthrax toxin that target host receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 13509–13513. [Google Scholar]
- Konig, R; Stertz, S; Zhou, Y; Inoue, A; Hoffmann, HH; Bhattacharyya, S; Alamares, JG; Tscherne, DM; Ortigoza, MB; Liang, Y; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar]
- Karlas, A; Machuy, N; Shin, Y; Pleissner, KP; Artarini, A; Heuer, D; Becker, D; Khalil, H; Ogilvie, LA; Hess, S; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar]
- Brass, AL; Dykxhoorn, DM; Benita, Y; Yan, N; Engelman, A; Xavier, RJ; Lieberman, J; Elledge, SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar]
- Zhou, H; Xu, M; Huang, Q; Gates, AT; Zhang, XD; Castle, JC; Stec, E; Ferrer, M; Strulovici, B; Hazuda, DJ; Espeseth, AS. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar]
- Borner, K; Hermle, J; Sommer, C; Brown, NP; Knapp, B; Glass, B; Kunkel, J; Torralba, G; Reymann, J; Beil, N; et al. From experimental setup to bioinformatics: An RNAi screening platform to identify host factors involved in HIV-1 replication. Biotechnol. J 2010, 5, 39–49. [Google Scholar]
- Pache, L; Konig, R; Chanda, SK. Identifying HIV-1 host cell factors by genome-scale RNAi screening. Methods 2011, 53, 3–12. [Google Scholar]
- Krishnan, MN; Ng, A; Sukumaran, B; Gilfoy, FD; Uchil, PD; Sultana, H; Brass, AL; Adametz, R; Tsui, M; Qian, F; et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar]
- Li, Q; Brass, AL; Ng, A; Hu, Z; Xavier, RJ; Liang, TJ; Elledge, SJ. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [Google Scholar]
- Sigoillot, FD; King, RW. Vigilance and Validation: Keys to Success in RNAi Screening. ACS Chem. Biol 2011, 6, 47–60. [Google Scholar]
- Huang, DW; Sherman, BT; Lempicki, RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009, 37, 1–13. [Google Scholar]
- Bard, JB; Rhee, SY. Ontologies in biology: design, applications and future challenges. Nat. Rev. Genet 2004, 5, 213–222. [Google Scholar]
- Rhee, SY; Wood, V; Dolinski, K; Draghici, S. Use and misuse of the gene ontology annotations. Nat. Rev. Genet 2008, 9, 509–515. [Google Scholar]
- Chuang, HY; Hofree, M; Ideker, T. A decade of systems biology. Annu. Rev. Cell Dev. Biol 2010, 26, 721–744. [Google Scholar]
- Jensen, LJ; Bork, P. Ontologies in quantitative biology: a basis for comparison, integration, and discovery. PLoS Biol 2010, 8, e1000374. [Google Scholar]
- Khatri, P; Draghici, S. Ontological analysis of gene expression data: Current tools, limitations, and open problems. Bioinformatics 2005, 21, 3587–3595. [Google Scholar]
- Ooi, HS; Schneider, G; Chan, YL; Lim, TT; Eisenhaber, B; Eisenhaber, F. Databases of protein-protein interactions and complexes. Methods Mol. Biol 2010, 609, 145–159. [Google Scholar]
- Ooi, HS; Schneider, G; Lim, TT; Chan, YL; Eisenhaber, B; Eisenhaber, F. Biomolecular pathway databases. Methods Mol. Biol 2010, 609, 129–144. [Google Scholar]
- Quackenbush, J. Computational analysis of microarray data. Nat. Rev. Genet 2001, 2, 418–427. [Google Scholar]
- Kanehisa, M; Goto, S; Furumichi, M; Tanabe, M; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 2010, 38, D355–D360. [Google Scholar]
- Kanehisa, M; Goto, S; Hattori, M; Aoki-Kinoshita, KF; Itoh, M; Kawashima, S; Katayama, T; Araki, M; Hirakawa, M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 2006, 34, D354–D357. [Google Scholar]
- Kanehisa, M; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000, 28, 27–30. [Google Scholar]
- Matthews, L; Gopinath, G; Gillespie, M; Caudy, M; Croft, D; de Bono, B; Garapati, P; Hemish, J; Hermjakob, H; Jassal, B; et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res 2009, 37, D619–D622. [Google Scholar]
- Vastrik, I; D’Eustachio, P; Schmidt, E; Gopinath, G; Croft, D; de Bono, B; Gillespie, M; Jassal, B; Lewis, S; Matthews, L; et al. Reactome: A knowledge base of biologic pathways and processes. Genome Biol 2007, 8, R39. [Google Scholar]
- Joshi-Tope, G; Vastrik, I; Gopinath, GR; Matthews, L; Schmidt, E; Gillespie, M; D’Eustachio, P; Jassal, B; Lewis, S; Wu, G; et al. The Genome Knowledgebase: A resource for biologists and bioinformaticists. Cold Spring Harb. Symp. Quant. Biol 2003, 68, 237–243. [Google Scholar]
- Thomas, PD; Campbell, MJ; Kejariwal, A; Mi, H; Karlak, B; Daverman, R; Diemer, K; Muruganujan, A; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res 2003, 13, 2129–2141. [Google Scholar]
- Thomas, PD; Kejariwal, A; Campbell, MJ; Mi, H; Diemer, K; Guo, N; Ladunga, I; Ulitsky-Lazareva, B; Muruganujan, A; Rabkin, S; et al. PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res 2003, 31, 334–341. [Google Scholar]
- Pico, AR; Kelder, T; van Iersel, MP; Hanspers, K; Conklin, BR; Evelo, C. WikiPathways: Pathway editing for the people. PLoS Biol 2008, 6, e184. [Google Scholar]
- Karp, PD; Ouzounis, CA; Moore-Kochlacs, C; Goldovsky, L; Kaipa, P; Ahren, D; Tsoka, S; Darzentas, N; Kunin, V; Lopez-Bigas, N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res 2005, 33, 6083–6089. [Google Scholar]
- Lee, SJ; Ways, JA; Barbato, JC; Essig, D; Pettee, K; DeRaedt, SJ; Yang, S; Weaver, DA; Koch, LG; Cicila, GT. Gene expression profiling of the left ventricles in a rat model of intrinsic aerobic running capacity. Physiol. Genomics 2005, 23, 62–71. [Google Scholar]
- Gene Ontology (GO). Available online: http://www.geneontology.org/ accessed on 9 June 2011.
- Szklarczyk, D; Franceschini, A; Kuhn, M; Simonovic, M; Roth, A; Minguez, P; Doerks, T; Stark, M; Muller, J; Bork, P; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011, 39, D561–D568. [Google Scholar]
- Jensen, LJ; Kuhn, M; Stark, M; Chaffron, S; Creevey, C; Muller, J; Doerks, T; Julien, P; Roth, A; Simonovic, M; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009, 37, D412–D416. [Google Scholar]
- von Mering, C; Jensen, LJ; Kuhn, M; Chaffron, S; Doerks, T; Kruger, B; Snel, B; Bork, P. STRING 7—recent developments in the integration and prediction of protein interactions. Nucleic Acids Res 2007, 35, D358–D362. [Google Scholar]
- von Mering, C; Jensen, LJ; Snel, B; Hooper, SD; Krupp, M; Foglierini, M; Jouffre, N; Huynen, MA; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res 2005, 33, D433–D437. [Google Scholar]
- von Mering, C; Huynen, M; Jaeggi, D; Schmidt, S; Bork, P; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res 2003, 31, 258–261. [Google Scholar]
- Snel, B; Lehmann, G; Bork, P; Huynen, MA. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res 2000, 28, 3442–3444. [Google Scholar]
- Kuhn, M; Szklarczyk, D; Franceschini, A; Campillos, M; von Mering, C; Jensen, LJ; Beyer, A; Bork, P. STITCH 2: An interaction network database for small molecules and proteins. Nucleic Acids Res 2010, 38, D552–D556. [Google Scholar]
- Kuhn, M; von Mering, C; Campillos, M; Jensen, LJ; Bork, P. STITCH: Interaction networks of chemicals and proteins. Nucleic Acids Res 2008, 36, D684–688. [Google Scholar]
- Dezso, Z; Nikolsky, Y; Nikolskaya, T; Miller, J; Cherba, D; Webb, C; Bugrim, A. Identifying disease-specific genes based on their topological significance in protein networks. BMC Syst Biol 2009, 3. [Google Scholar] [CrossRef]
- LaCount, DJ; Vignali, M; Chettier, R; Phansalkar, A; Bell, R; Hesselberth, JR; Schoenfeld, LW; Ota, I; Sahasrabudhe, S; Kurschner, C; et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 2005, 438, 103–107. [Google Scholar]
- Bader, GD; Hogue, CW. BIND—a data specification for storing and describing biomolecular interactions, molecular complexes and pathways. Bioinformatics 2000, 16, 465–477. [Google Scholar]
- Bader, GD; Donaldson, I; Wolting, C; Ouellette, BF; Pawson, T; Hogue, CW. BIND—The Biomolecular Interaction Network Database. Nucleic Acids Res 2001, 29, 242–245. [Google Scholar]
- Ceol, A; Chatr Aryamontri, A; Licata, L; Peluso, D; Briganti, L; Perfetto, L; Castagnoli, L; Cesareni, G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res 2010, 38, D532–D539. [Google Scholar]
- Prasad, TS; Kandasamy, K; Pandey, A. Human protein reference database and human proteinpedia as discovery tools for systems biology. Methods Mol. Biol 2009, 577, 67–79. [Google Scholar]
- Mishra, GR; Suresh, M; Kumaran, K; Kannabiran, N; Suresh, S; Bala, P; Shivakumar, K; Anuradha, N; Reddy, R; Raghavan, TM; et al. Human protein reference database—2006 update. Nucleic Acids Res 2006, 34, D411–D414. [Google Scholar]
- Peri, S; Navarro, JD; Amanchy, R; Kristiansen, TZ; Jonnalagadda, CK; Surendranath, V; Niranjan, V; Muthusamy, B; Gandhi, TK; Gronborg, M; et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res 2003, 13, 2363–2371. [Google Scholar]
- Diez, D; Wheelock, AM; Goto, S; Haeggstrom, JZ; Paulsson-Berne, G; Hansson, GK; Hedin, U; Gabrielsen, A; Wheelock, CE. The use of network analyses for elucidating mechanisms in cardiovascular disease. Mol. Biosyst 2010, 6, 289–304. [Google Scholar]
- Tarca, AL; Draghici, S; Khatri, P; Hassan, SS; Mittal, P; Kim, JS; Kim, CJ; Kusanovic, JP; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar]
- Qu, XA; Gudivada, RC; Jegga, AG; Neumann, EK; Aronow, BJ. Inferring novel disease indications for known drugs by semantically linking drug action and disease mechanism relationships. BMC Bioinformatics 2009, 10. [Google Scholar] [CrossRef]
- Sthoeger, ZM; Zinger, H; Mozes, E. Beneficial effects of the anti-oestrogen tamoxifen on systemic lupus erythematosus of (NZBxNZW)F1 female mice are associated with specific reduction of IgG3 autoantibodies. Ann. Rheum. Dis 2003, 62, 341–346. [Google Scholar]
- Cohen-Solal, JF; Jeganathan, V; Grimaldi, CM; Peeva, E; Diamond, B. Sex hormones and SLE: influencing the fate of autoreactive B cells. Curr. Top. Microbiol. Immunol 2006, 305, 67–88. [Google Scholar]
- Wishart, DS; Knox, C; Guo, AC; Cheng, D; Shrivastava, S; Tzur, D; Gautam, B; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 2008, 36, D901–D906. [Google Scholar]
- Wishart, DS; Knox, C; Guo, AC; Shrivastava, S; Hassanali, M; Stothard, P; Chang, Z; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006, 34, D668–D672. [Google Scholar]
- Kellenberger, E; Muller, P; Schalon, C; Bret, G; Foata, N; Rognan, D. sc-PDB: An annotated database of druggable binding sites from the Protein Data Bank. J. Chem. Inf. Model 2006, 46, 717–727. [Google Scholar]
- Berman, HM; Bhat, TN; Bourne, PE; Feng, Z; Gilliland, G; Weissig, H; Westbrook, J. The Protein Data Bank and the challenge of structural genomics. Nat. Struct. Biol 2000, 7, 957–959. [Google Scholar]
- Berman, HM; Westbrook, J; Feng, Z; Gilliland, G; Bhat, TN; Weissig, H; Shindyalov, IN; Bourne, PE. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar]
- Li, Q; Cheng, T; Wang, Y; Bryant, SH. PubChem as a public resource for drug discovery. Drug Discov. Today 2010, 15, 1052–1057. [Google Scholar]
- Olah, M; Rad, R; Ostopovici, L; Bora, A; Hadaruga, N; Hadaruga, R; Moldovan, R; Fulias, A; Mracec, M; Opera, TI. Schreiber, SL, Kapoor, TM, Wess, G, Eds.; WOMBAT and WOMBAT-PK: Bioactivity Databases for Lead and Drug Discovery. In Chemical Biology: From Small Molecules to Systems Biology and Drug Design; Wiley-VCH: New York, NY, USA, 2007; pp. 760–786. [Google Scholar]
- Opera, TI; Benedetti, P; Berellini, G; Olah, M; Fejgin, K; Boyer, S. Cruciani, G, Ed.; Rapid ADME Filters for Lead Discovery. In Molecular Interactions Field; Wiley-VCH: New York, NY, USA, 2006; pp. 249–272. [Google Scholar]
- Scheer, M; Grote, A; Chang, A; Schomburg, I; Munaretto, C; Rother, M; Sohngen, C; Stelzer, M; Thiele, J; Schomburg, D. BRENDA, the enzyme information system in 2011. Nucleic Acids Res 2011, 39, D670–D676. [Google Scholar]
- Shoichet, BK. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar]
- Konig, R; Zhou, YY; Elleder, D; Diamond, TL; Bonamy, GMC; Irelan, JT; Chiang, CY; Tu, BP; De Jesus, PD; Lilley, CE; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar]
- Bushman, FD; Malani, N; Fernandes, J; D’Orso, I; Cagney, G; Diamond, TL; Zhou, HL; Hazuda, DJ; Espeseth, AS; Konig, R; et al. Host Cell Factors in HIV Replication: Meta-Analysis of Genome-Wide Studies. PLoS Pathog 2009, 5, e1000437. [Google Scholar]
- Barrows, NJ; Le Sommer, C; Garcia-Blanco, MA; Pearson, JL. Factors affecting reproducibility between genome-scale siRNA-based screens. J. Biomol. Screen 2010, 15, 735–747. [Google Scholar]
- Gene Ontology Consortium. The Gene Ontology in 2010: Extensions and refinements. Nucleic Acids Res 2010, 38, D331–D335.
- The Reference Genome Group of the Gene Ontology Consortium. The Gene Ontology’s Reference Genome Project: A unified framework for functional annotation across species. PLoS Comput. Biol 2009, 5, e1000431.
- Dennis, G, Jr; Sherman, BT; Hosack, DA; Yang, J; Gao, W; Lane, HC; Lempicki, RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003, 4. [Google Scholar]
- Fu, W; Sanders-Beer, BE; Katz, KS; Maglott, DR; Pruitt, KD; Ptak, RG. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res 2009, 37, D417–D422. [Google Scholar]
- Garrus, JE; von Schwedler, UK; Pornillos, OW; Morham, SG; Zavitz, KH; Wang, HE; Wettstein, DA; Stray, KM; Cote, M; Rich, RL; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar]
- Kilzer, JM; Stracker, T; Beitzel, B; Meek, K; Weitzman, M; Bushman, FD. Roles of host cell factors in circularization of retroviral dna. Virology 2003, 314, 460–467. [Google Scholar]
- Wei, P; Garber, ME; Fang, SM; Fischer, WH; Jones, KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar]
- Yedavalli, VS; Neuveut, C; Chi, YH; Kleiman, L; Jeang, KT. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 2004, 119, 381–392. [Google Scholar]
- Fellay, J; Shianna, KV; Ge, D; Colombo, S; Ledergerber, B; Weale, M; Zhang, K; Gumbs, C; Castagna, A; Cossarizza, A; et al. A whole-genome association study of major determinants for host control of HIV-1. Science 2007, 317, 944–947. [Google Scholar]
- Matthews, PC; Prendergast, A; Leslie, A; Crawford, H; Payne, R; Rousseau, C; Rolland, M; Honeyborne, I; Carlson, J; Kadie, C; et al. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. J. Virol 2008, 82, 8548–8559. [Google Scholar]
- Cherepanov, P. LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic Acids Res 2007, 35, 113–124. [Google Scholar]
- Cherepanov, P; Maertens, G; Proost, P; Devreese, B; Van Beeumen, J; Engelborghs, Y; De Clercq, E; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem 2003, 278, 372–381. [Google Scholar]
- Ciuffi, A; Bushman, FD. Retroviral DNA integration: HIV and the role of LEDGF/p75. Trends Genet 2006, 22, 388–395. [Google Scholar]
- Gamble, TR; Vajdos, FF; Yoo, S; Worthylake, DK; Houseweart, M; Sundquist, WI; Hill, CP. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar]
- Llano, M; Saenz, DT; Meehan, A; Wongthida, P; Peretz, M; Walker, WH; Teo, W; Poeschla, EM. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [Google Scholar]
- Maertens, G; Cherepanov, P; Pluymers, W; Busschots, K; De Clercq, E; Debyser, Z; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem 2003, 278, 33528–33539. [Google Scholar]
- Marshall, HM; Ronen, K; Berry, C; Llano, M; Sutherland, H; Saenz, D; Bickmore, W; Poeschla, E; Bushman, FD. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2007, 2, e1340. [Google Scholar]
- Turlure, F; Devroe, E; Silver, PA; Engelman, A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci 2004, 9, 3187–3208. [Google Scholar]
- Ciuffi, A; Llano, M; Poeschla, E; Hoffmann, C; Leipzig, J; Shinn, P; Ecker, JR; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med 2005, 11, 1287–1289. [Google Scholar]
- Llano, M; Vanegas, M; Fregoso, O; Saenz, D; Chung, S; Peretz, M; Poeschla, EM. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol 2004, 78, 9524–9537. [Google Scholar]
- Hao, L; Sakurai, A; Watanabe, T; Sorensen, E; Nidom, CA; Newton, MA; Ahlquist, P; Kawaoka, Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008, 454, 890–893. [Google Scholar]
- Brass, AL; Huang, IC; Benita, Y; John, SP; Krishnan, MN; Feeley, EM; Ryan, BJ; Weyer, JL; van der Weyden, L; Fikrig, E; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar]
- Shapira, SD; Gat-Viks, I; Shum, BO; Dricot, A; de Grace, MM; Wu, L; Gupta, PB; Hao, T; Silver, SJ; Root, DE; et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [Google Scholar]
- Josset, L; Textoris, J; Loriod, B; Ferraris, O; Moules, V; Lina, B; N’Guyen, C; Diaz, JJ; Rosa-Calatrava, M. Gene expression signature-based screening identifies new broadly effective influenza a antivirals. PLoS One 2010, 5, e13169. [Google Scholar]
- Coombs, KM; Berard, A; Xu, W; Krokhin, O; Meng, X; Cortens, JP; Kobasa, D; Wilkins, J; Brown, EG. Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J. Virol 2010, 84, 10888–10906. [Google Scholar]
- Watanabe, T; Watanabe, S; Kawaoka, Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 2010, 7, 427–439. [Google Scholar]
- Min, JY; Subbarao, K. Cellular targets for influenza drugs. Nat. Biotechnol 2010, 28, 239–240. [Google Scholar]
- Chase, G; Deng, T; Fodor, E; Leung, BW; Mayer, D; Schwemmle, M; Brownlee, G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 2008, 377, 431–439. [Google Scholar]
- Gonzalez, O; Fontanes, V; Raychaudhuri, S; Loo, R; Loo, J; Arumugaswami, V; Sun, R; Dasgupta, A; French, SW. The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology 2009, 50, 1756–1764. [Google Scholar]
- Soh, D; Dong, D; Guo, Y; Wong, L. Consistency, comprehensiveness, and compatibility of pathway databases. BMC Bioinformatics 2010, 11, 449F. [Google Scholar]
- Ingenuity Systems. Available online: http://www.ingenuity.com/index.html accessed on 6 January 2011.
- Konig, R; Stertz, S; Zhou, Y; Inoue, A; Hoffmann, HH; Bhattacharyya, S; Alamares, JG; Tscherne, DM; Ortigoza, MB; Liang, Y; Gao, Q; Andrews, SE; Bandyopadhyay, S; De Jesus, P; Tu, BP; Pache, L; Shih, C; Orth, A; Bonamy, G; Miraglia, L; Ideker, T; Garcia-Sastre, A; Young, JA; Palese, P; Shaw, ML; Chanda, SK. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar]
- Karlas, A; Machuy, N; Shin, Y; Pleissner, KP; Artarini, A; Heuer, D; Becker, D; Khalil, H; Ogilvie, LA; Hess, S; Maurer, AP; Muller, E; Wolff, T; Rudel, T; Meyer, TF. Genomewide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar]
- Brass, AL; Huang, IC; Benita, Y; John, SP; Krishnan, MN; Feeley, EM; Ryan, BJ; Weyer, JL; van der Weyden, L; Fikrig, E; Adams, DJ; Xavier, RJ; Farzan, M; Elledge, SJ. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar]
- Shapira, SD; Gat-Viks, I; Shum, BO; Dricot, A; de Grace, MM; Wu, L; Gupta, PB; Hao, T; Silver, SJ; Root, DE; Hill, DE; Regev, A; Hacohen, N. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [Google Scholar]
- Josset, L; Textoris, J; Loriod, B; Ferraris, O; Moules, V; Lina, B; N’Guyen, C; Diaz, JJ; Rosa-Calatrava, M. Gene expression signature-based screening identifies new broadly effective influenza a antivirals. PLoS One 2010, 5. [Google Scholar]
- Chen, YL; Chen, WW; Wang, YF; Li, RL; Guo, WF; Lao, SX; Wang, JH; Huang, SP. Bioinformatics research on chronic superficial gastritis of Pi-deficiency syndrome by gene arrays. Chin. J. Integr. Med 2009, 15, 341–346. [Google Scholar]






| Database | Description | References |
|---|---|---|
| Kyoto Encyclopedia of Genes and Genome (KEGG) | Public Resource links genes to crystal structures and drugs when information is available | [52–54] |
| Reactome | Public Resource accepts a gene list for the pathway analyzer and returns percentage population per pathway | [55–57] |
| Protein Analysis Through Evolutionary Relationships (PANTHER) | Free pathway database allows user to identify enrichment in biological pathways, GO terms or protein class | [58,59] |
| WikiPathways | Community curated pathway database | [60,61] |
| Ingenuity IPA | Commercial pathway database to identify enrichment in pathways/GO terms; links drugs to specific genes | [62] |
| Gene Ontology (GO) Consortium | Community database that clusters genes by biological process, molecular function or cellular location across multiple species | [63] |
| Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) | Freely available functional relationships database displays direct neighborhood relationships between proteins that interact directly or through an intermediary | [64–69] |
| Search Tool for Interactions of Chemicals (STITCH) | Crosslinks gene products with chemical structures from PubChem | [70,71] |
| GeneGo Metacore | Commercial manually curated pathway database annotated with 600,000 compounds | [72] |
| Prolexys HyNet | Commercial database protein-protein interaction identified via in-house yeast two-hybrid screening | [73] |
| Biomolecular Interaction Network Database (BIND) | Free and Commercial versions describing protein-protein interactions, molecular complexes and pathways | [74,75] |
| Molecular Interactions Database (MINT) | Public protein-protein interaction database based on peerreviewed literature. Accessible through web-interface or Simple Object Access Protocol/Representational State Transfer (SOAP/REST) protocols | [76] |
| Human Protein Reference Database (HPRD) | Public proteonomic database with descriptions for 2750 human proteins taken from the primary literature | [77–79] |
| GeneGO/MCODE | STRING | PANTHER | Ingenuity IPA | Reactome |
|---|---|---|---|---|
| Translation Initiation | Translation Initiation | Apoptosis signaling pathway | Chronic Myeloid Leukemia Signaling | Dissolution of Fibrin Clot |
| Pre-mRNA Processing | Pre-mRNA Processing | T cell activation | B Cell Receptor Signaling | Influenza Life Cycle |
| Proton-Transporter V-type ATPase | Proton-Transporter V-type ATPase | Angiogenesis | Production of Nitric Oxide and Reactive Oxygen Species in Macrophages | MAP kinase cascade |
| COPI coating of Golgi vesicle | Toll receptor signaling | EIF2 Signaling | Metabolism of nitric oxide | |
| Nuclear Transport | Inflammation mediated by chemokine and cytokine signaling pathways | Rank Signaling in Osteoclast | Eukaryotic Translation Initiation | |
| Cell cycle | CD40 Signaling | Signaling by FGFR | ||
| PDGF signaling | Molecular Mechanisms of Cancer | Eukaryotic Translation Termination | ||
| FGF signaling | Role of PKR in Interferon Induction and Antiviral Response | Eukaryotic Translation Elongation | ||
| FAS signaling | Regulation of beta-cell development | |||
| Ras Pathway | Signaling by Insulin receptor | |||
| B cell activation | Processing of Capped Intron-Containing Pre-mRNA |
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Prussia, A.; Thepchatri, P.; Snyder, J.P.; Plemper, R.K. Systematic Approaches towards the Development of Host-Directed Antiviral Therapeutics. Int. J. Mol. Sci. 2011, 12, 4027-4052. https://doi.org/10.3390/ijms12064027
Prussia A, Thepchatri P, Snyder JP, Plemper RK. Systematic Approaches towards the Development of Host-Directed Antiviral Therapeutics. International Journal of Molecular Sciences. 2011; 12(6):4027-4052. https://doi.org/10.3390/ijms12064027
Chicago/Turabian StylePrussia, Andrew, Pahk Thepchatri, James P. Snyder, and Richard K. Plemper. 2011. "Systematic Approaches towards the Development of Host-Directed Antiviral Therapeutics" International Journal of Molecular Sciences 12, no. 6: 4027-4052. https://doi.org/10.3390/ijms12064027
APA StylePrussia, A., Thepchatri, P., Snyder, J. P., & Plemper, R. K. (2011). Systematic Approaches towards the Development of Host-Directed Antiviral Therapeutics. International Journal of Molecular Sciences, 12(6), 4027-4052. https://doi.org/10.3390/ijms12064027