Solvolyses of Benzoyl Chlorides in Weakly Nucleophilic Media

Abstract
:1. Introduction
2. Results and Discussion
2.1. Reliability of Rate Constants
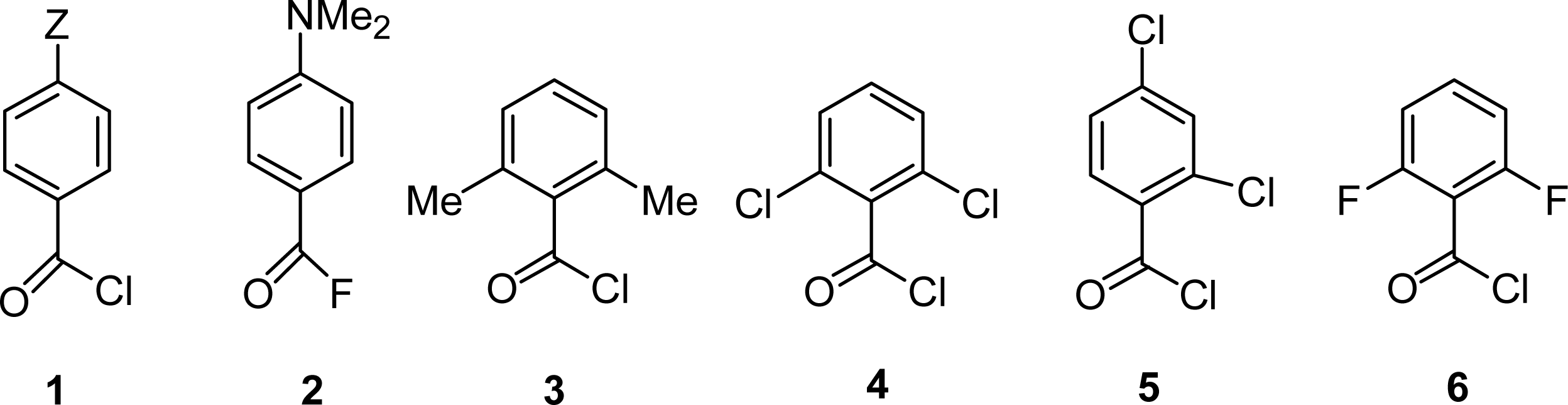
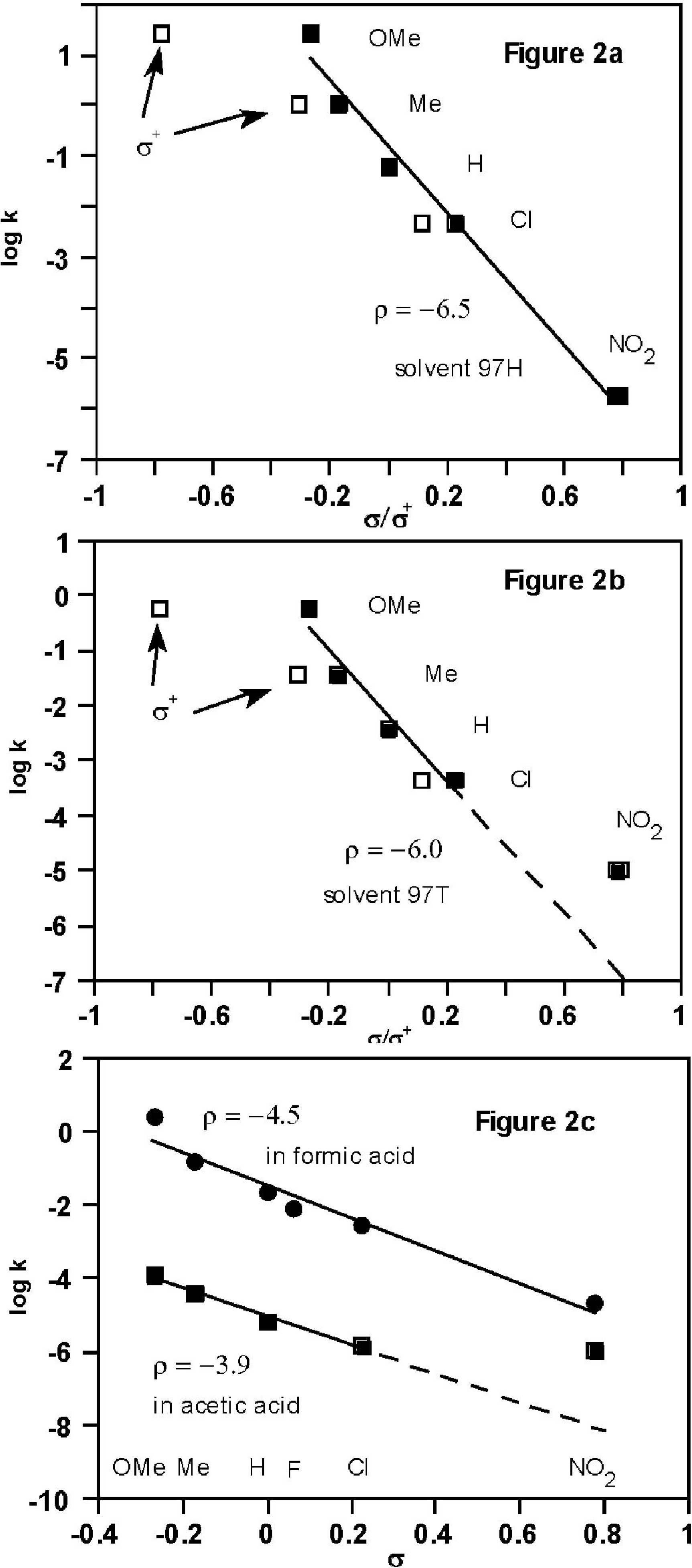
2.2. Kinetic Data (Substituent Effects)
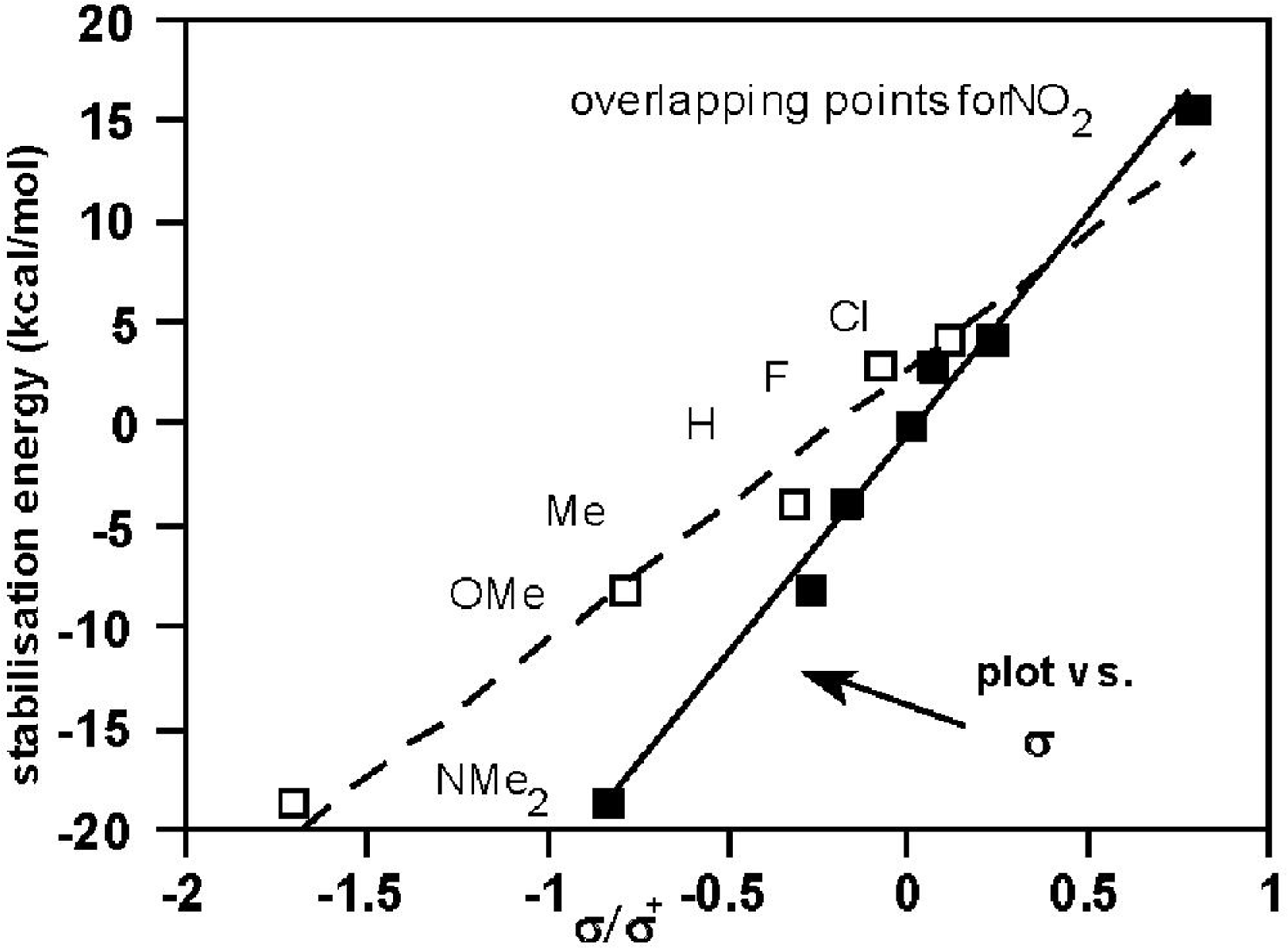
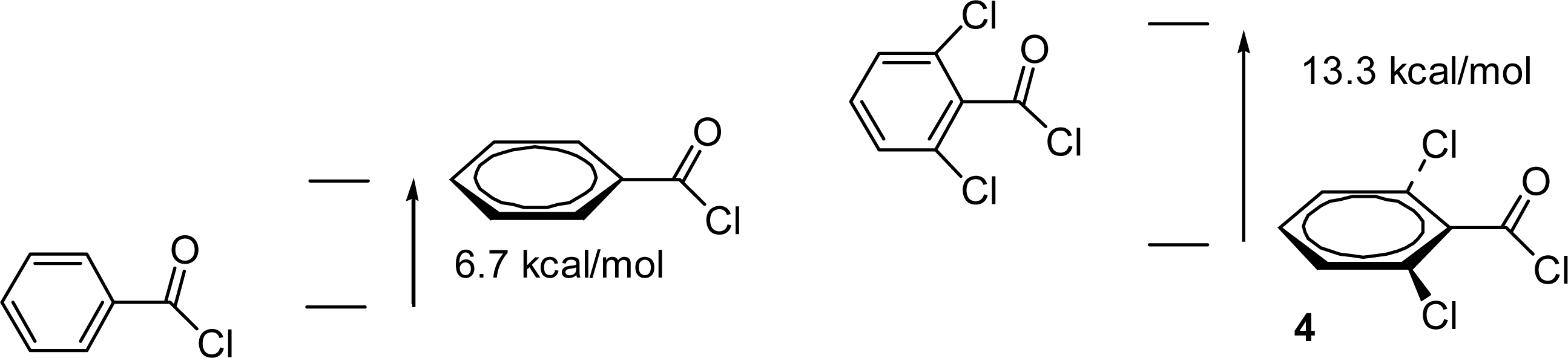
2.3. Theoretical Calculations of Substituent Effects

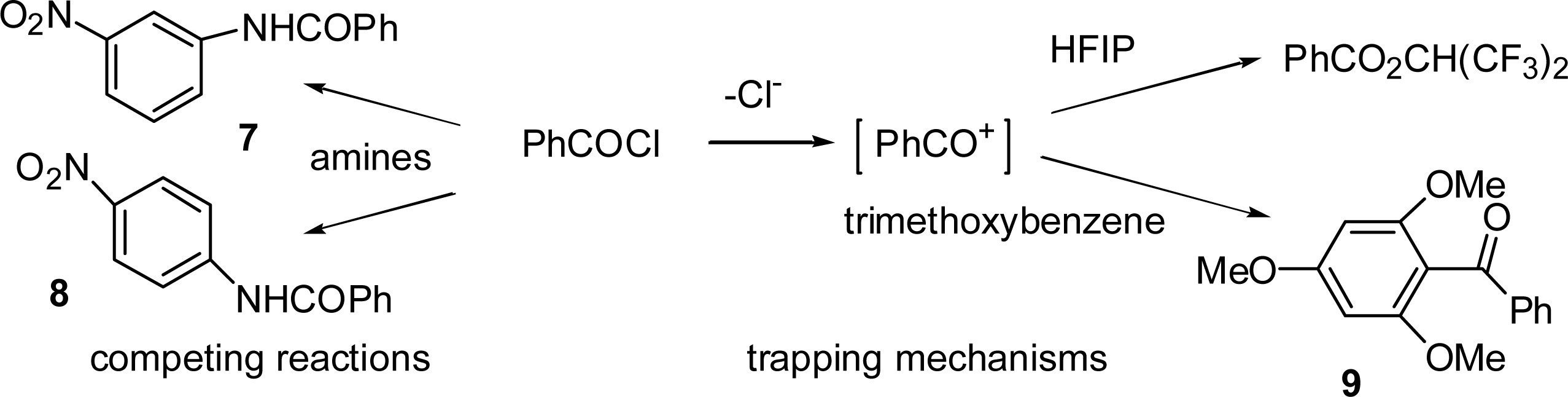
2.4. Product Studies and Reaction Mechanisms
3. Experimental Section
4. Conclusions
Acknowledgments
References and Notes
- Bentley, TW; Harris, HC. Solvolyses of para-substituted benzoyl chlorides in trifluoroethanol and in highly aqueous media. J Chem Soc, Perkin Trans 1986, 2, 619–624. [Google Scholar] [CrossRef]
- Bentley, TW; Koo, IS; Norman, SJ. Distinguishing between solvation effects and mechanistic changes. Effects due to differences in solvation between aromatic rings and alkyl groups. J. Org. Chem 1991, 56, 1604–1609. [Google Scholar]
- Song, BD; Jencks, WP. Mechanisms of solvolysis of substituted benzoyl halides. J. Am. Chem. Soc 1989, 111, 8470–8479. [Google Scholar]
- Bender, ML; Chen, MC. Acylium ion formation in the reactions of carboxylic acid chlorides. III. The hydrolysis of 4-substituted-2,6-dimethylbenzoyl chlorides. J. Am. Chem. Soc 1963, 85, 30–36. [Google Scholar]
- Bentley, TW; Harris, HC; Koo, IS. Rapid solvolyses of 2,6-dimethyl- and 2,4,6-trimethyl-benzoyl chlorides: Model systems for solvent effects on the reactivity of acid chlorides. J Chem Soc, Perkin Trans 1988, 2, 783–789. [Google Scholar] [CrossRef]
- Park, KH; Kevill, DN. Influence of the ortho-effect in the solvolyses of 2,6-dichlorobenzoyl chlorides. J Phys Org Chem 2011. [Google Scholar] [CrossRef]
- Kevill, DN; D’Souza, MJ. Correlation of the rates of solvolysis of benzoyl chloride and derivatives using extended forms of the Grunwald-Winstein equation. J. Phys. Org. Chem 2002, 15, 881–888. [Google Scholar]
- Park, KH; Kevill, DN. The importance of the ortho effect in the solvolyses of 2,6-difluorobenzoyl chloride. J Phys Org Chem 2011. [Google Scholar] [CrossRef]
- García-Río, L; Hall, RW; Mejuto, JC; Rodriguez-Defonte, P. The solvolysis of benzoyl halides as a chemical probe determining the polarity of the cavity of dimethyl-β-cyclodextrin. Tetrahedron 2007, 63, 2208–2214. [Google Scholar]
- Bentley, TW; Jones, RO. Stoichiometric solvation effects. Part 1. New equations relating product selectivities to alcohol-water solvent compositions for hydrolyses of p-nitrobenzoyl chloride. J Chem Soc, Perkin Trans 1993, 2, 2351–2357. [Google Scholar]
- D’Souza, MJ; Kevill, DN; Bentley, TW; Devaney, AC. Kinetics and selectivities for solvolysis of N,N-diphenylcarbamoyl chloride. J. Org. Chem 1996, 60, 1632–1637. [Google Scholar]
- Kevill, DN; Rudolph, TM; D’Souza, MJ. Solvolyis of N,N-dimethylthiocarbamoyl chloride; effect of sulfur-for-oxygen substitution upon kinetics and product partitioning. J. Phys. Org. Chem 2000, 13, 192–196. [Google Scholar]
- Bentley, TW; Carter, GE; Harris, HC. Competing SN2 and carbonyl addition pathways for solvolyses of benzoyl chloride in aqueous media. J Chem Soc, Perkin Trans 1985, 2, 983–990. [Google Scholar] [CrossRef]
- Fainberg, AH; Winstein, S. Correlation of solvolysis rates. III. t-butyl chloride in a wide range of solvent mixtures. J. Am. Chem. Soc 1956, 78, 2770–2777. [Google Scholar]
- Swain, CG; Mosely, RB; Bown, DE. Correlation of rates of solvolysis with a four-parameter equation. J. Am. Chem. Soc 1955, 77, 3731–3737. [Google Scholar]
- Crunden, EW; Hudson, RF. The mechanism of hydrolysis of acid chlorides. Part V1. Formolysis of para-substituted benzoyl chlorides. J Chem Soc 1956, 501–507. [Google Scholar]
- Johnson, SL. General base and nucleophilic catalysis of ester hydrolysis and related reactions. Adv. Phys. Org. Chem 1967, 5, 237–330. [Google Scholar]
- Liu, KT; Chen, HI. Solvent and substituent effects in solvolyses of benzoyl chlorides. Variation of mechanisms from Grunwald-Winstein correlation analyses with YBnCl scales. J Chem Soc, Perkin Trans 2000, 2, 893–898. [Google Scholar] [CrossRef]
- Brown, HC; Okamoto, Y. Electrophilic substituent constants. J. Am. Chem. Soc 1958, 80, 4979–4987. [Google Scholar]
- Nakata, K; Fujio, M; Nishimito, K; Tsuno, Y. Theoretical studies on empirical structure-reactivity relationship: the Yukawa-Tsuno equation. J. Phys. Org. Chem 2003, 16, 323–335. [Google Scholar]
- The predictions require k0 = 4.49 × 10−3 s−1 for 1, Z = Cl in 80% ethanol-water at 25 °C [22].
- Lee, I; Koo, IS; Sohn, SC; Lee, HH. Nucleophilic substitution at a carbonyl carbon. 14. Transition state variation in the solvolysis of benzoyl chlorides. Bull. Korean Chem. Soc 1982, 3, 92–98. [Google Scholar]
- Traeger, JC. Heat of formation of the benzoyl cation by photoionization mass spectrometry. Eur. J. Mass Spectrom 2009, 15, 183–188. [Google Scholar]
- Bentley, TW. Structural effects on the solvolytic reactivity of carboxylic and sulfonic acid chlorides. Comparisons with gas phase data for cation formation. J. Org. Chem 2008, 73, 6251–6257. [Google Scholar]
- Bentley, TW; Jones, RO. Substituent effects on the formation of sulfonyl cations from sulfonyl chlorides: comparisons of solvolysis kinetic data with calculated gas phase energies. J. Phys. Org. Chem 2007, 20, 1093–1101. [Google Scholar]
- Krygowski, TM; Cyrański, MK; Sung, DD; Stepień, BT. Solvolysis of aromatic benzoyl chlorides: How is the π-electron stabilization of the aromatic acyl chlorides and acylium cations related to the π-electron delocalization? J. Phys. Org. Chem 2004, 17, 699–706. [Google Scholar]
- Liu, KT; Chen, PS; Chiu, PF; Tsao, ML. Electronic and steric effects on the solvation at the transition-state in the solvolysis of some tertiary benzylic halides. Tetrahedron Lett 1992, 33, 6499–6502. [Google Scholar]
- Hofmann, M; Hampel, N; Kanzian, T; Mayr, H. Electrophilic alkylations in neutral aqueous or alcoholic solutions. Angew. Chem. Int. Ed 2004, 43, 5402–5405. [Google Scholar]
- Bentley, TW; Ebdon, DN; Kim, EJ; Koo, IS. Solvent polarity and organic reactivity in mixed solvents: evidence using a molecular probe to assess the role of preferential solvation in aqueous alcohols. J. Org. Chem 2005, 70, 1647–1653. [Google Scholar]
- Bentley, TW; Carter, GE; Harris, HC. SN2 character of hydrolysis of benzoyl chloride. J Chem Soc Chem Comm 1984, 387–389. [Google Scholar] [CrossRef]
- Bentley, TW; Bowen, CT; Morten, DH; Schleyer, PVR. The SN2-SN1 spectrum 3. Solvolyses of secondary and tertiary alkyl sulfonates in fluorinated alcohols. Further evidence for the SN2 intermediate mechanism. J. Am. Chem. Soc 1981, 103, 5466–5475. [Google Scholar]
- It has been proposed [18], based on comparisons with 3 that solvolyses of 1, Z = OMe involve nucleophilic solvent assistance (NSA), so the calculations based on 1, Z = OMe are minimum estimates of NSA.
- Schadt, FL; Bentley, TW; Schleyer, PvR. The SN2-SN1 spectrum 2. Quantitative treatments of nucleophilic solvent assistance. A scale of solvent nucleophilicities. J. Am. Chem. Soc 1976, 98, 7667–7674. [Google Scholar]
- Lambert, JB; Putz, GJ; Mixan, CE. Stereochemistry of the solvolysis of cyclohexyl tosylate. J. Am. Chem. Soc 1972, 94, 5132–5133. [Google Scholar]
- Bentley, TW; Llewellyn, G; McAlister, JA. SN2 mechanism for alcoholysis, aminolysis and hydrolysis of acetyl chloride. J. Org. Chem 1996, 61, 7927–7932. [Google Scholar]
- D’Souza, MJ; Ryu, ZH; Park, BC; Kevill, DN. Correlation of rates of solvolysis of acetyl chloride and α-substituted derivatives. Can. J. Chem 2008, 86, 359–367. [Google Scholar]
- Regan, A; Watt, CIF. Structure and reactivity in the hydrolyses of carboxylic acid esters and chlorides. J. Phys. Org. Chem 2007, 20, 180–189. [Google Scholar]
- Ruff, F; Farkas, O. Concerted SN2 mechanism for the hydrolysis of acid chlorides: comparisons of reactivities calculated by density functional theory with experimental data. J. Phys. Org. Chem 2011, 24, 480–491. [Google Scholar]
- Bentley, TW; Freeman, AE. Mechanistic applications of high performance liquid chromatography. Rate-product correlations for competing solvolysis and aminolysis of benzoyl chloride. J Chem Soc, Perkin Trans 1984, 2, 1115–1119. [Google Scholar] [CrossRef]
- Ciamician, G; Silber, P. Synthese des benzophloroglucintrimethyläther. Berichte der Deutschen Chemischen Gesellschaft 1894, 27, 1497–1501. [Google Scholar]
- Bentley, TW; Carter, GE; Roberts, K. Solvent ionizing power. Comparisons of solvolyses of 1-adamantyl chlorides, bromides, iodides, and tosylates in protic solvents. J. Org. Chem 1984, 49, 5183–5189. [Google Scholar]
- Gaussian 03; Revision D.01; Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA, Jr; Vreven, T; Kudin, KN; Burant, JC; et al. Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate b | Temperature/°C | k/s−1 | ΔH≠/kcal mol−1 | ΔS≠/cal mol−1 K−1 |
|---|---|---|---|---|
| 1, Z = OMe | −20.15 | (3.15 ± 0.03) × 10−1 | 14.2 | −4.3 |
| −9.90 | (9.87 ± 0.02) × 10−1 | |||
| 25.0 c | 27 | |||
| 1, Z = Me | −10.0 d | (4.08 ± 0.06) × 10−2 | 14.0 | −11.4 |
| 0.1 | (1.14 ± 0.01) × 10−1 | |||
| 25.0 c | 1.07 | |||
| 1, Z = H | 0.0 d | (5.45 ± 0.13) × 10−3 | 14.9 | −14.2 |
| 10.0 | (1.49 ± 0.02) × 10−2 | |||
| 25.0 c,e | 5.95 × 10−2 | |||
| 1, Z = Cl | 5.1 | (6.67 ± 0.07) × 10−4 | 15.5 | −17.2 |
| 25.0 f | (4.65 ± 0.01) × 10−3 | |||
| 25.0 f,g | (4.54 ± 0.07) × 10−3 | |||
| 1, Z = NO2 | 25.0 h | (1.77 ± 0.08) × 10−6 |
| Substrate b | Temperature/°C | k/s−1 | ΔH≠/kcal mol−1 | ΔS≠/cal mol−1 K−1 |
|---|---|---|---|---|
| 1, Z = OMe | 5.0 d | (2.53 ± 0.21) × 10−1 | 18.0 | 3.4 |
| 10.0 | (4.57 ± 0.10) × 10−1 | |||
| 25.0 c | 2.4 |
| Substrate | Acetic acid | Note b | Formic acid | Note b |
|---|---|---|---|---|
| 1, Z = OMe | (1.23 ± 0.11) × 10−4 | UV c,d | 2.4 | Table 2 |
| (1.18 ± 0.07) × 10−4 | Titr | |||
| 1, Z = Me | 3.98 × 10−5 | [15] | (1.53 ± 0.03) × 10−1 | Cond c |
| 1, Z = H | (6.7 ± 0.3) × 10−6 | Titr e | (2.11 ± 0.14) × 10−2 | Cond f,g |
| 1, Z = F | (7.3 ± 0.2) × 10−3 | Cond c | ||
| 1, Z = Cl | (1.42 ± 0.12) × 10−6 | Titr | (2.74 ± 0.16) × 10−3 | Cond h,i |
| (1.71 ± 0.05) × 10−3 | UV h,j | |||
| 1, Z = NO2 | 1.05 × 10−6 | [15] | 2.09 × 10−5 | [15] |
| Substrate | E(ArCOCl)/Hartrees b | E(ArCO+)/Hartrees b | Stabilisation energies/kcal mol−1 | HBDE/kcal mol−1 | ||
|---|---|---|---|---|---|---|
| HF/6–31G(d) | B3LYP/6–31G(d) | 6–311G(d,p) | ||||
| 1, Z = H | −802.34371 c | −342.59639 c | 0.0 d | 0.0 d | 0.0 d | 150.1 e |
| 1, Z = F | −901.19505 | −441.44325 | 2.8 | 1.6 | 2.07 f | 152.3 |
| 1, Z = Cl | −1261.24202 c | −801.48796 c | 4.2 | 2.3 | 2.11 f | 153.3 |
| 1, Z = NO2 | −1005.80911 g | −546.03707 g | 15.5 | 12.05 f | 163.9 | |
| 1, Z = NMe2 | −935.42722 | −475.70965 | −18.7h | −20.2h | 130.7h | |
| 1, Z = OMe | −916.22720 c | −456.49301 c | −8.2 | −9.6 | −9.54 f | 141.2 |
| 1, Z = Me | −841.38219 g | −381.64104 g | −3.9 | −4.54 f | 145.9 | |
| 2,6-diMe (3) | −880.40699 c | −420.68210 c | −14.1 h | −14.5 h | 135.8 h | |
| 2,6-diCl (4) | −1720.12086 | −1260.37391 | −0.2 | −2.7 | −1.8 | 148.9 |
| 2,4-diCl (5) | −1720.12652 | −1260.37638 | 1.8 | −0.1 | 151.0 | |
| 3,4-diCl | −1720.13317 | −1260.37130 | 9.1 | 6.0 | 157.6 | |
| 3,5-diCl | −1720.13634 | −1260.37021 | 11.8 | 9.5 | 160.8 | |
| 2,6-diF (6) | −1000.03037 | −540.28117 | 1.2 | 0.0 | 150.7 | |
| 3-OMe i | −916.22245 | −456.47692 | −1.1 | −3.3 | 147.9 | |
| Substrate a | k/s−1 (55 °C) b | k/s−1 (25 °C) | log kobs c | log kcalc d | Δlog k e |
|---|---|---|---|---|---|
| 3,4-dichloro | 1.40 × 10−3 | 1.12 × 10−4 | −3.92 | −3.7 | 0.2 |
| 3,5-dichloro | 1.35 × 10−4 | 7.43 × 10−6 b | −5.13 | −4.7 | 0.4 |
| 2,6-diMe (3) | 3.9 f | 3.2 | −0.7 | ||
| 2,6-dichoro (4) | 5.62 × 10−2 | 5.6 × 10−3 g | −2.25 | −0.95 | 1.3 |
| 2,4-dichloro (5) | 1.51 × 10−3 | 1.5 × 10−4 g | −3.8 | −1.6 | 2.2 |
| 2,6-difluoro (6) | 3.46 × 10−3 h | 3.5 × 10−4 g | −3.45 | −1.5 | 1.95 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bentley, T.W.; Harris, H.C. Solvolyses of Benzoyl Chlorides in Weakly Nucleophilic Media. Int. J. Mol. Sci. 2011, 12, 4805-4818. https://doi.org/10.3390/ijms12084805
Bentley TW, Harris HC. Solvolyses of Benzoyl Chlorides in Weakly Nucleophilic Media. International Journal of Molecular Sciences. 2011; 12(8):4805-4818. https://doi.org/10.3390/ijms12084805
Chicago/Turabian StyleBentley, Thomas William, and Haldon Carl Harris. 2011. "Solvolyses of Benzoyl Chlorides in Weakly Nucleophilic Media" International Journal of Molecular Sciences 12, no. 8: 4805-4818. https://doi.org/10.3390/ijms12084805
APA StyleBentley, T. W., & Harris, H. C. (2011). Solvolyses of Benzoyl Chlorides in Weakly Nucleophilic Media. International Journal of Molecular Sciences, 12(8), 4805-4818. https://doi.org/10.3390/ijms12084805