High-Density Real-Time PCR-Based in Vivo Toxicogenomic Screen to Predict Organ-Specific Toxicity


Abstract
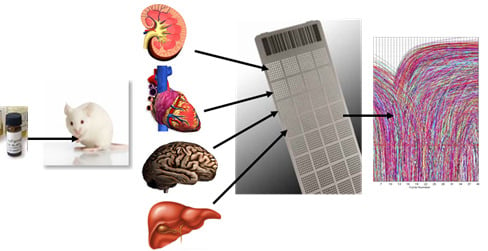
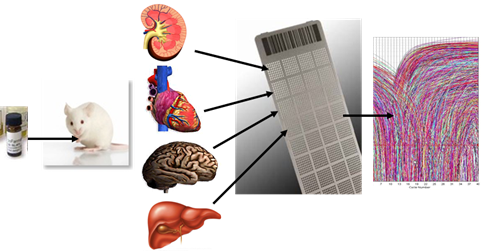
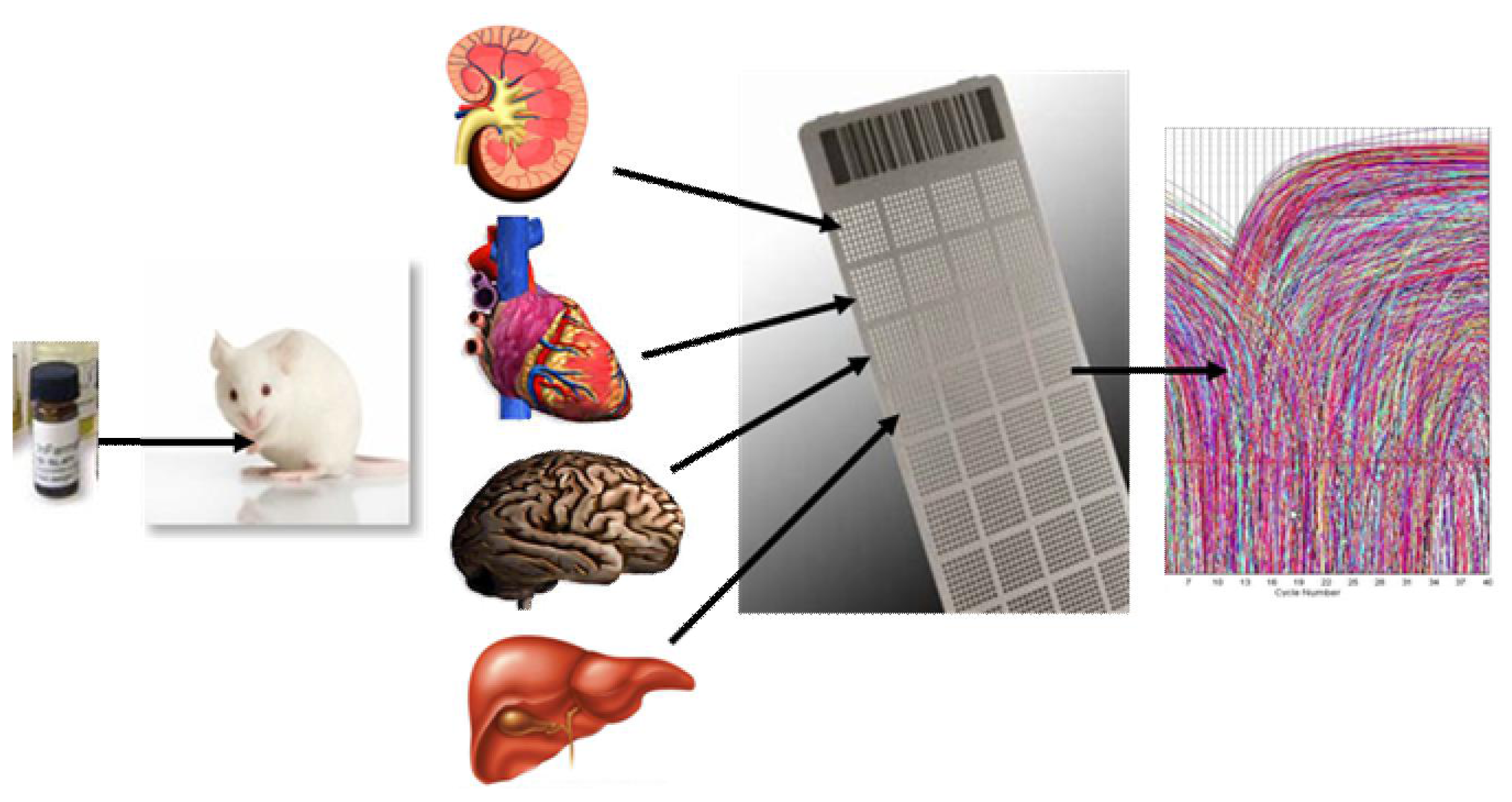
:
1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Development of a Toxicogenomic Nanocapillary QRT-PCR Platform
2.1.2. In Vivo Protocol for Toxicogenomic Profiling of Multiple Organs
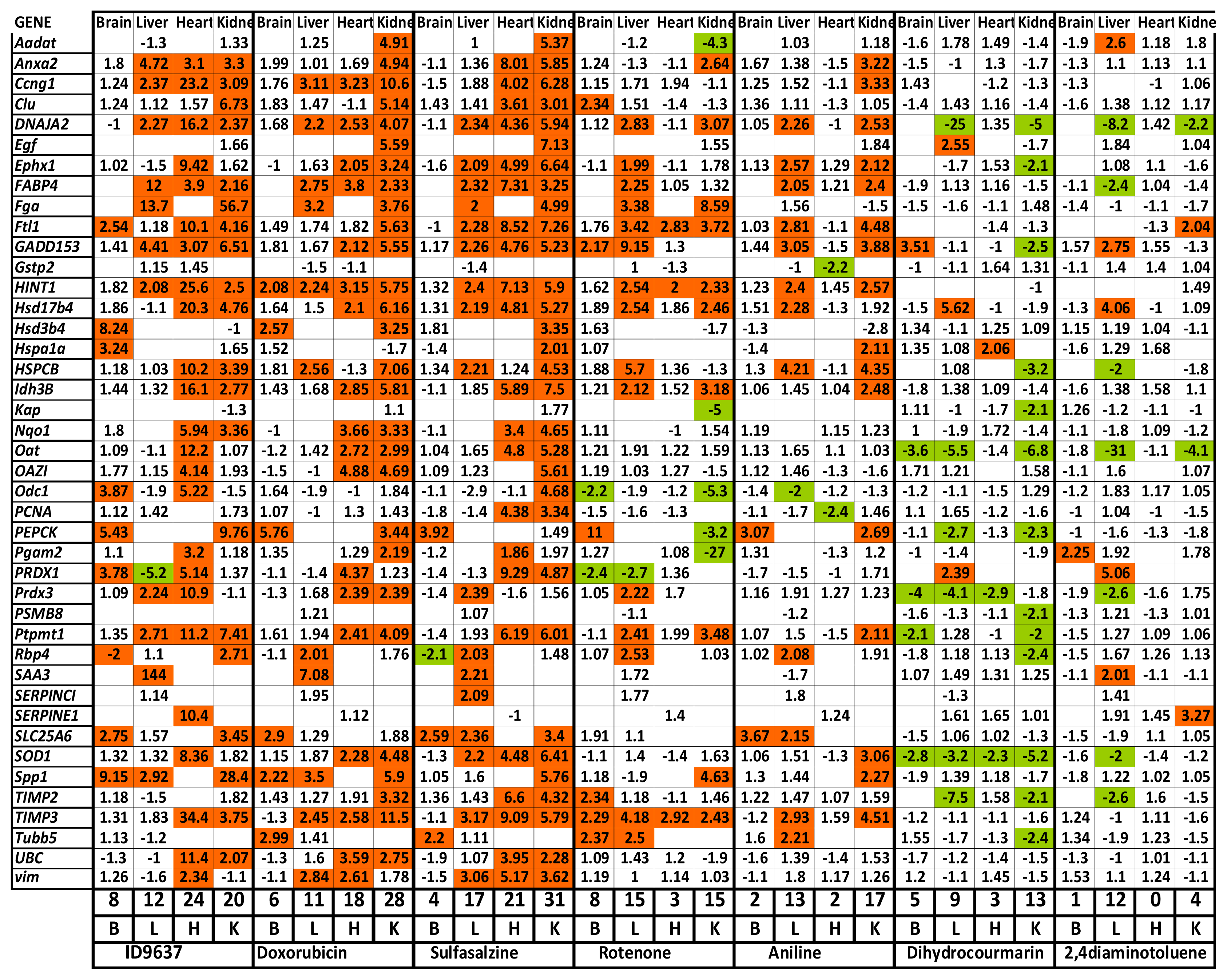
2.1.3. Profiling of Known Toxic Reference Compounds
2.1.4. Profiling of Drugs and Prodrugs
2.1.5. Comparison of Data from Toxicogenomic and Histological Analysis
2.2. Discussion
3. Experimental Section
3.1. Animals, Treatment, and Sample Collection
3.2. RNA Isolation
3.3. Profiling of RNAs with High-Throughput, Nanocapillary QRT-PCR
4. Conclusions
Supplementary Information
ijms-12-06116-s001.zipAcknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
Abbreviations
| QRT-PCR | quantitative real-time PCR |
| ADME | absorption, distribution, metabolism and excretion |
References
- Waring, JF; Halbert, DN. The promise of toxicogenomics. Curr Opin Mol Ther 2002, 4, 229–235. [Google Scholar]
- Vass, L; Kelemen, JZ; Fehér, LZ; Lorincz, Z; Kulin, S; Cseh, S; Dormán, G; Puskás, LG. Toxicogenomics screening of small molecules using high-density, nanocapillary real-time PCR. Int J Mol Med 2009, 23, 65–74. [Google Scholar]
- Lord, PG; Nie, A; McMillian, M. Application of genomics in preclinical drug safety evaluation. Basic Clin Pharmacol Toxicol 2006, 98, 537–546. [Google Scholar]
- Van Hummelen, P; Sasaki, J. State-of-the-art genomics approaches in toxicology. Mutat Res 2010, 705, 165–171. [Google Scholar]
- Jayapal, M; Bhattacharjee, RN; Melendez, AJ; Hande, MP. Environmental toxicogenomics: A post-genomic approach to analysing biological responses to environmental toxins. Int J Biochem Cell Biol 2010, 42, 230–240. [Google Scholar]
- Dai, X; He, YD; Dai, H; Lum, PY; Roberts, CJ; Waring, JF; Ulrich, RG. Development of an approach for ab initio estimation of compound-induced liver injury based on global gene transcriptional profiles. Genome Inform 2006, 17, 77–88. [Google Scholar]
- Hamadeh, HK; Bushel, PR; Jayadev, S; Martin, K; DiSorbo, O; Sieber, S; Bennett, L; Tennant, R; Stoll, R; Barrett, JC; Blanchard, K; Paules, RS; Afshari, CA. Gene expression analysis reveals chemical-specific profiles. Toxicol Sci 2002, 67, 219–231. [Google Scholar]
- Burczynski, ME; McMillian, M; Ciervo, J; Li, L; Parker, JB; Dunn, RT, II; Hicken, S; Farr, S; Johnson, MD. Toxicogenomics-based discrimination of toxic mechanism in HepG2 human hepatoma cells. Toxicol Sci 2000, 58, 399–415. [Google Scholar]
- Kier, LD; Neft, R; Tang, L; Suizu, R; Cook, T; Onsurez, K; Tiegler, K; Sakai, Y; Ortiz, M; Nolan, T; Sankar, U; Li, AP. Applications of microarrays with toxicologically relevant genes (tox genes) for the evaluation of chemical toxicants in Sprague Dawley rats in vivo and human hepatocytesin vitro. Mutat Res 2004, 549, 101–113. [Google Scholar]
- Glass, KY; Newsome, CR; Tchounwou, PB. Cytotoxicity and expression of c-fos, HSP70, and GADD45/153 proteins in human liver carcinoma (HepG2) cells exposed to dinitrotoluenes. Int J Environ Res Public Health 2005, 2, 355–361. [Google Scholar]
- Franc, MA; Moffat, ID; Boutros, PC; Tuomisto, JT; Tuomisto, J; Pohjanvirta, R; Okey, AB. Patterns of dioxin-altered mRNA expression in livers of dioxin-sensitive versus dioxin-resistant rats. Arch Toxicol 2008, 82, 809–830. [Google Scholar]
- Wang, EJ; Snyder, RD; Fielden, MR; Smith, RJ; Gu, YZ. Validation of putative genomic biomarkers of nephrotoxicity in rats. Toxicology 2008, 246, 91–100. [Google Scholar]
- Rokushima, M; Omi, K; Imura, K; Araki, A; Furukawa, N; Itoh, F; Miyazaki, M; Yamamoto, J; Rokushima, M; Okada, M; Torii, M; Kato, I; Ishizaki, J. Toxicogenomics of drug-induced hemolytic anemia by analyzing gene expression profiles in the spleen. Toxicol Sci 2007, 100, 290–302. [Google Scholar]
- Malard, V; Berenguer, F; Prat, O; Ruat, S; Steinmetz, G; Quemeneur, E. Global gene expression profiling in human lung cells exposed to cobalt. BMC Genomics 2007, 8, 147. [Google Scholar]
- Dam, K; Seidler, FJ; Slotkin, TA. Transcriptional biomarkers distinguish between vulnerable periods for developmental neurotoxicity of chlorpyrifos: Implications for toxicogenomics. Brain Res Bull 2003, 59, 261–265. [Google Scholar]
- Ellinger-Ziegelbauer, H; Aubrecht, J; Kleinjans, JC; Ahr, HJ. Application of toxicogenomics to study mechanisms of genotoxicity and carcinogenicity. Toxicol Lett 2009, 186, 36–44. [Google Scholar]
- Sawada, H; Takami, K; Asahi, S. A toxicogenomic approach to drug-induced phospholipidosis: analysis of its induction mechanism and establishment of a novel in vitro screening system. Toxicol Sci 2005, 83, 282–292. [Google Scholar]
- Akilesh, S; Shaffer, DJ; Roopenian, D. Customized molecular phenotyping by quantitative gene expression and pattern recognition analysis. Genome Res 2003, 13, 1719–1727. [Google Scholar]
- Rohrbeck, A; Salinas, G; Maaser, K; Linge, J; Salovaara, S; Corvi, R; Borlak, J. Toxicogenomics applied to in vitro carcinogenicity testing with Balb/c 3T3 cells revealed a gene signature predictive of chemical carcinogens. Toxicol Sci 2010, 118, 31–41. [Google Scholar]
- Catalá, A; Zvara, A; Puskás, LG; Kitajka, K. Melatonin-induced gene expression changes and its preventive effects on adriamycin-induced lipid peroxidation in rat liver. J Pineal Res 2007, 42, 43–49. [Google Scholar]
- Kiyosawa, N; Manabe, S; Yamoto, T; Sanbuissho, A. Practical application of toxicogenomics for profiling toxicant-induced biological perturbations. Int J Mol Sci 2010, 11, 3397–3412. [Google Scholar]
- Glass, KY; Newsome, CR; Tchounwou, PB. Cytotoxicity and expression of c-fos, HSP70, and GADD45/153 proteins in human liver carcinoma (HepG2) cells exposed to dinitrotoluenes. Int J Environ Res Public Health 2005, 2, 355–361. [Google Scholar]
- Walsh, PJ; Bookman, R; Zaias, J; Mayer, GD; Abraham, W; Bourdelais, AJ; Baden, DG. Toxicogenomic effects of marine brevetoxins in liver and brain of mouse. Comp Biochem Physiol B Biochem Mol Biol 2003, 136, 173–182. [Google Scholar]
- Seth, D; Hogg, PJ; Gorrell, MD; McCaughan, GW; Haber, PS. Direct effects of alcohol on hepatic fibrinolytic balance: implications for alcoholic liver disease. J Hepatol 2008, 48, 614–627. [Google Scholar]
- Petrov, AV. Assessment of sulfasalazine and hydroxichloroqine hepatotoxicity in patients with rheumatic arthritis and isolated HBS-antigen positivity. Lik Sprava 2004, 1, 60–65. [Google Scholar]
- Cavard, C; Terris, B; Grimber, G; Christa, L; Audard, V; Radenen-Bussiere, B; Simon, MT; Renard, CA; Buendia, MA; Perret, C. Overexpression of regenerating islet-derived 1 alpha and 3 alpha genes in human primary liver tumors with beta-catenin mutations. Oncogene 2006, 25, 599–608. [Google Scholar]
- Berthiaume, JM; Wallace, KB. Persistent alterations to the gene expression profile of the heart subsequent to chronic Doxorubicin treatment. Cardiovasc Toxicol 2007, 7, 178–191. [Google Scholar]
- Yamamoto, K; Loskutoff, DJ. The kidneys of mice with autoimmune disease acquire a hypofibrinolytic/procoagulant state that correlates with the development of glomerulonephritis and tissue microthrombosis. Am J Pathol 1997, 151, 725–734. [Google Scholar]
- Zhang, H; Shi, Z; Liu, Y; Wei, Y; Dai, J. Lipid homeostasis and oxidative stress in the liver of male rats exposed to perfluorododecanoic acid. Toxicol Appl Pharmacol 2008, 227, 16–25. [Google Scholar]
- Kinser, S; Jia, Q; Li, M; Laughter, A; Cornwell, P; Corton, JC; Pestka, J. Gene expression profiling in spleens of deoxynivalenol-exposed mice: immediate early genes as primary targets. J Toxicol Environ Health A 2004, 67, 1423–1441. [Google Scholar]
- Tohyama, M; Shirakata, Y; Sayama, K; Hashimoto, K. The influence of hepatic damage on serum soluble Fas ligand levels of patients with drug rashes. J Allergy Clin Immunol 2009, 123, 971–972. [Google Scholar]
- Bulera, SJ; Eddy, SM; Ferguson, E; Jatkoe, TA; Reindel, JF; Bleavins, MR; De La Iglesia, FA. RNA expression in the early characterization of hepatotoxicants in Wistar rats by high-density DNA microarrays. Hepatology 2001, 33, 1239–1258. [Google Scholar]
- Combs, AB; Acosta, D. Toxic mechanisms of the heart: A review. Toxicol Pathol 1990, 18, 583–596. [Google Scholar]
- Lapointe, N; St-Hilaire, M; Martinoli, MG; Blanchet, J; Gould, P; Rouillard, C; Cicchetti, F. Rotenone induces non-specific central nervous system and systemic toxicity. FASEB J 2004, 18, 717–719. [Google Scholar]
- Séverin, I; Jondeau, A; Dahbi, L; Chagnon, MC. 2,4-Diaminotoluene (2,4-DAT)-induced DNA damage, DNA repair and micronucleus formation in the human hepatoma cell line HepG2. Toxicology 2005, 213, 138–146. [Google Scholar]
- Puskás, LG; Fehér, LZ; Vizler, C; Ayaydin, F; Rásó, E; Molnár, E; Magyary, I; Kanizsai, I; Gyuris, M; Madácsi, R; Fábián, G; Farkas, K; Hegyi, P; Baska, F; Ozsvári, B; Kitajka, K. Polyunsaturated fatty acids synergize with lipid droplet binding thalidomide analogs to induce oxidative stress in cancer cells. Lipids Health Dis 2010, 9, 56. [Google Scholar]
- Szabó, A; Répási, J; Fábián, G; Tiszlavicz, L; Puskás, LG; Ózsvári, B. Ubichem Ltd., Avidin Ltd; unpublished work; 2010.
- Curran, MP; Wagstaff, AJ. Estradiol and norgestimate: A review of their combined use as hormone replacement therapy in postmenopausal women. Drugs Aging 2001, 18, 863–885. [Google Scholar]
- Johnson, DE; Wolfgang, GH. Predicting human safety: Screening and computational approaches. Drug Discov Today 2000, 5, 445–454. [Google Scholar]
- Giri, S; Nieber, K; Bader, A. Hepatotoxicity and hepatic metabolism of available drugs: Current problems and possible solutions in preclinical stages. Expert Opin Drug Metab Toxicol 2010, 6, 895–917. [Google Scholar]
- Khan, KNM; Alden, CL. Kidney. In Handbook of Toxicological Pathology, 2nd ed; Haschek, WM, Rousseaux, CG, Wallig, MA, Eds.; Academic Press: San Diego, CA, USA, 2001; pp. 255–330. [Google Scholar]
- Linares, V; Alonso, V; Domingo, JL. Oxidative stress as a mechanism underlying sulfasalazine-induced toxicity. Expert Opin Drug Saf 2011, 10, 253–263. [Google Scholar]
- Boiteux, G; Lascombe, I; Roche, E; Plissonnier, ML; Clairotte, A; Bittard, H; Fauconnet, S. A-FABP, a candidate progression marker of human transitional cell carcinoma of the bladder, is differentially regulated by PPAR in urothelial cancer cells. Int J Cancer 2009, 124, 1820–1828. [Google Scholar]
- Cabré, A; Lázaro, I; Girona, J; Manzanares, JM; Marimón, F; Plana, N; Heras, M; Masana, L. Fatty acid binding protein 4 is increased in metabolic syndrome and with thiazolidinedione treatment in diabetic patients. Atherosclerosis 2007, 195, e150–e158. [Google Scholar]
- Ordovas, JM. Genetic links between diabetes mellitus and coronary atherosclerosis. Curr Atheroscler Rep 2007, 9, 204–210. [Google Scholar]
- Blandizzi, C; De Paolis, B; Colucci, R; Di Paolo, A; Danesi, R; Del Tacca, M. Acetylcholinesterase blockade does not account for the adverse cardiovascular effects of the antitumor drug irinotecan: a preclinical study. Toxicol Appl Pharmacol 2001, 177, 149–156. [Google Scholar]
- de Jong, FA; van der Bol, JM; Mathijssen, RH; van Gelder, T; Wiemer, EA; Sparreboom, A; Verweij, J. Renal function as a predictor of irinotecan-induced neutropenia. Clin Pharmacol Ther 2008, 84, 254–262. [Google Scholar]
- Faragó, N; Zvara, Á; Varga, Z; Ferdinandy, P; Puskás, LG. Purification of high-quality microRNA from the heart tissue. Acta Biol Hung 62. in press.



{kind=link}
{kind=link}
{kind=link}
| # | Gene | Name | Accession No. | Probe Name | Organ | Ref. |
|---|---|---|---|---|---|---|
| 1 | GADD153 | DNA-damage-inducible 3 | NM_007837.3 | Mm00492097_m1 | liver | [7,21] |
| 2 | SAA3 | serum amyloid A 3 | NM_011315.3 | Mm00441203_m1 | liver, lung | [22] |
| 3 | TIMP3 | metallopeptidase inhibitor 3 | NM_011595.2 | Mm00441827_m1 | liver, lung | [22] |
| 4 | PEPCK | phosphoenolpyr. carboxykinase | NM_011044.2 | Mm00440636_m1 | liver | [5] |
| 5 | NOX3 | NADPH oxidase 3 | NM_198958.2 | Mm01339132_m1 | kidney | [6] |
| 6 | Hsd3b4 | hydroxy-d-5-steroid dehyd. | NM_001111336 | Mm00843753_s1 | liver | [7] |
| 7 | Clu | clusterin | NM_013492.2 | Mm00442773_m1 | kidney, liver | [7,11] |
| 8 | Spp1 | secreted phosphoprotein 1 | NM_001204201 | Mm00436767_m1 | kidney | [11] |
| 9 | vim | vimentin | NM_011701 | Mm01333430_m1 | kidney | [11] |
| 10 | Anxa2 | annexin A2 | NM_007585.3 | Mm00500307_m1 | kidney | [23] |
| 11 | Tubb5 | tubulin, beta 5 | NM_011655.5 | Mm00495804_m1 | kidney | [11] |
| 12 | Gstp2 | glutathione S-transferase, pi 2 | NM_181796.2 | Mm00839138_g1 | kidney | [7] |
| 13 | Fga | fibrinogen alpha chain | NM_001111048 | Mm00802584_m1 | kidney | [24] |
| 14 | Ccng1 | cyclin G1 | NM_009831.2 | Mm00438084_m1 | kidney | [11] |
| 15 | Klk1b3 | kallikrein 1-related peptidase b3 | NM_008693.2 | Mm01203825_gH | kidney | [11] |
| 16 | Odc1 | ornithine decarboxylase 1 | NM_013614.2 | Mm01964631_g1 | kidney | [11] |
| 17 | Kap | kidney androgen regulated prot. | NM_010594.2 | Mm00495104_m1 | kidney | [11] |
| 18 | Oat | ornithine aminotransferase | NM_016978.2 | Mm00497544_m1 | kidney | [11] |
| 19 | Rbp4 | retinol binding protein 4 | NM_001159487 | Mm00803266_m1 | kidney | [11] |
| 20 | Aadat | aminoadipate aminotransferase | NM_011834.2 | Mm00496169_m1 | kidney | [11] |
| 21 | Egf | epidermal growth factor | NM_010113.3 | Mm01316968_m1 | kidney | [11] |
| 22 | Pgam2 | phosphoglycerate mutase 2 | NM_018870.3 | Mm00450782_g1 | heart | [25] |
| 23 | Hsd17b4 | hydroxysteroid dehydrogenase 4 | NM_008292.4 | Mm00500443_m1 | heart | [25] |
| 24 | Idh3B | isocitrate dehydrogenase 3 beta | NM_130884.4 | Mm00504589_m1 | heart | [25] |
| 25 | Ndufa5 | NADH dehydrogenase 1 alpha 5 | NM_026614.2 | Mm00471676_g1 | heart | [25] |
| 26 | Prdx3 | peroxiredoxin 3 | NM_007452.2 | Mm00545848_m1 | heart | [25] |
| 27 | Alox12b | arachidonate 12-lipoxygenase | NM_009659.2 | Mm00507782_m1 | heart, brain | [26] |
| 28 | Reg3a | regenerating islet-derived 3a | NM_011259.1 | Mm00441121_m1 | heart, liver | [27] |
| 29 | Cyp1a1 | cytochrome P450, family 1a1 | NM_001136059 | Mm00487218_m1 | liver | [7] |
| 30 | SERPINE1 | serine peptidase inhibitor E1 | NM_008871.2 | Mm00435860_m1 | heart, kidney | [28] |
| 31 | CYP7A1 | cytochrome P450, family 7a1 | NM_007824.2 | Mm00484152_m1 | heart, liver | [29] |
| 32 | Akr1b8 | aldo-keto reductase family 1B8 | NM_008012.1 | Mm00484314_m1 | spleen | [12] |
| 33 | FABP4 | fatty acid binding protein 4 | NM_024406.2 | Mm00445878_m1 | spleen | [12] |
| 34 | Ptpmt1 | protein tyrosine phosphatase 1 | NM_025576.2 | Mm00458631_m1 | spleen | [30] |
| 35 | HINT1 | histidine triad nucl. binding prot. | NM_008248.2 | Mm00801722_m1 | spleen | [30] |
| 36 | PSMB8 | proteasome subunit, beta 8 | NM_010724.2 | Mm00440207_m1 | spleen | [30] |
| 37 | Hoxa2 | homeobox A2 | NM_010451.1 | Mm00439361_m1 | brain | [22] |
| 38 | DNAJA2 | DnaJ (Hsp40) homolog,A2 | NM_019794.4 | Mm00444898_m1 | lung, liver | [7,13] |
| 39 | OAZI | antizyme inhibitor 1 | NM_001102458 | Mm00497630_m1 | lung | [13] |
| 40 | SLC25A6 | solute carrier family 25A6 | NM_026255.5 | Mm00470958_m1 | lung | [13] |
| 41 | SERPINCI | serpin peptidase inhibitor, C1 | NM_000488.3 | Mm00446573_m1 | lung | [13] |
| 42 | HSPCB | heat shock protein 90 alpha B1 | NM_008302.3 | Mm00833431_g1 | lung | [13] |
| 43 | UBC | ubiquitin C | NM_019639.4 | Mm01201237_m1 | lung | [13] |
| 44 | TIMP2 | tissue inhib. metalloprot. 2 | NM_011594.3 | Mm00441825_m1 | lung | [13] |
| 45 | FAS | Fas (TNF receptor superfamily 6) | NM_001146708 | Mm01204974_m1 | liver | [31,32] |
| 46 | PCNA | proliferating cell nuclear antigen | NM_011045.2 | Mm00448100_g1 | liver | [7,11] |
| 47 | PRDX1 | peroxiredoxin 1 | NM_011034.4 | Mm01621996_s1 | liver, lung | [7] |
| 48 | Ephx1 | epoxide hydrolase 1 | NM_010145.2 | Mm00468752_m1 | spleen | [7] |
| 49 | Hspa1a | heat shock protein 1A | NM_010479.2 | Mm01159846_s1 | liver | [7] |
| 50 | SOD1 | superoxide dismutase 1 | NM_011434.1 | Mm01344233_g1 | liver, heart | [7] |
| 51 | Ftl1 | ferritin light chain 1 | NM_010240.2 | Mm03030144_g1 | liver, spleen | [12] |
| 52 | Nqo1 | NAD(P)H dehydrogenase 1 | NM_008706.5 | Mm00500821_m1 | liver | [7] |
| 53 | c-Fos | FBJ osteosarcoma oncogene | NM_010234.2 | Mm00487425_m1 | liver | [7,21] |
| 54 | PPIA | peptidylprolyl isomerase A | NM_008907.1 | Mm02342430_g1 | control | - |
| 55 | PGK1 | phosphoglycerate kinase 1 | NM_000291.3 | Mm00435617_m1 | control | - |
| 56 | RPLP0 | ribosomal protein, large, P0 | NM_007475.5 | Mm00725448_s1 | control | - |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fabian, G.; Farago, N.; Feher, L.Z.; Nagy, L.I.; Kulin, S.; Kitajka, K.; Bito, T.; Tubak, V.; Katona, R.L.; Tiszlavicz, L.; et al. High-Density Real-Time PCR-Based in Vivo Toxicogenomic Screen to Predict Organ-Specific Toxicity. Int. J. Mol. Sci. 2011, 12, 6116-6134. https://doi.org/10.3390/ijms12096116
Fabian G, Farago N, Feher LZ, Nagy LI, Kulin S, Kitajka K, Bito T, Tubak V, Katona RL, Tiszlavicz L, et al. High-Density Real-Time PCR-Based in Vivo Toxicogenomic Screen to Predict Organ-Specific Toxicity. International Journal of Molecular Sciences. 2011; 12(9):6116-6134. https://doi.org/10.3390/ijms12096116
Chicago/Turabian StyleFabian, Gabriella, Nora Farago, Liliana Z. Feher, Lajos I. Nagy, Sandor Kulin, Klara Kitajka, Tamas Bito, Vilmos Tubak, Robert L. Katona, Laszlo Tiszlavicz, and et al. 2011. "High-Density Real-Time PCR-Based in Vivo Toxicogenomic Screen to Predict Organ-Specific Toxicity" International Journal of Molecular Sciences 12, no. 9: 6116-6134. https://doi.org/10.3390/ijms12096116
APA StyleFabian, G., Farago, N., Feher, L. Z., Nagy, L. I., Kulin, S., Kitajka, K., Bito, T., Tubak, V., Katona, R. L., Tiszlavicz, L., & Puskas, L. G. (2011). High-Density Real-Time PCR-Based in Vivo Toxicogenomic Screen to Predict Organ-Specific Toxicity. International Journal of Molecular Sciences, 12(9), 6116-6134. https://doi.org/10.3390/ijms12096116