Protein Arginine Methyltransferases (PRMTs): Promising Targets for the Treatment of Pulmonary Disorders



{kind=link}
{kind=link}
Abstract
:1. Introduction
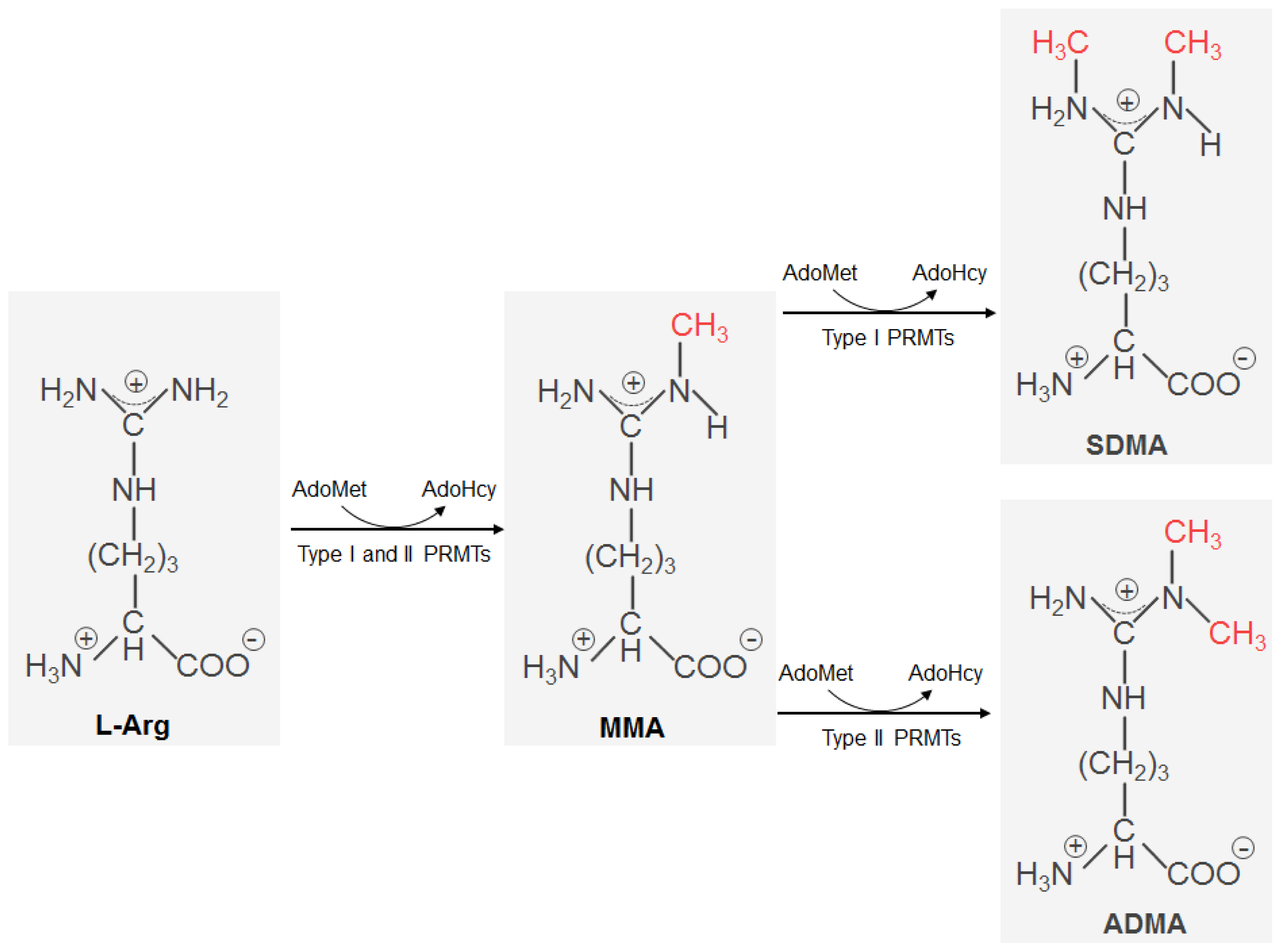
2. Protein Arginine Methylation
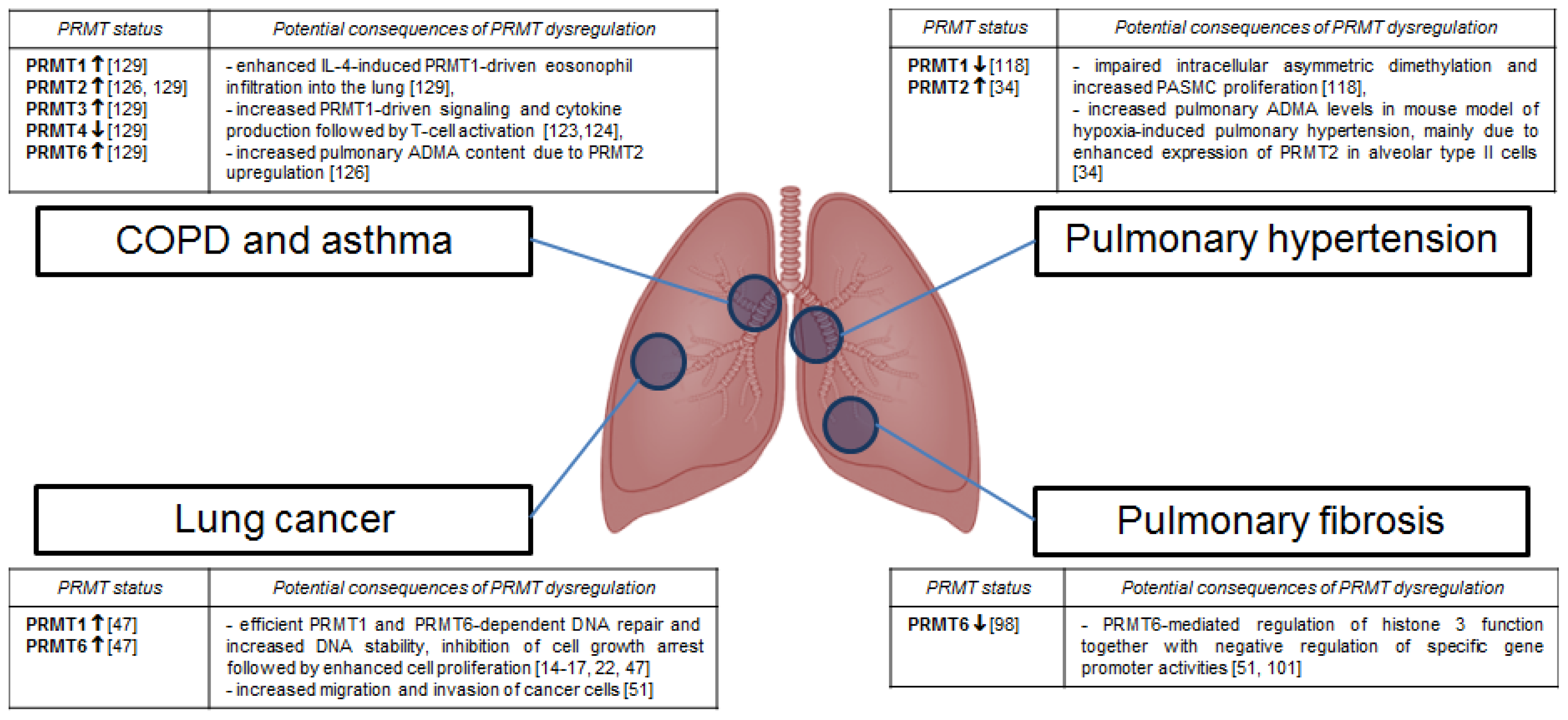
3. Protein Arginine Methyltransferases in Pulmonary Disorders
3.1. Lung Cancer
3.2. Pulmonary Fibrosis
3.3. Pulmonary Hypertension
3.4. Chronic Obstructive Pulmonary Disease and Asthma
4. Conclusions
Acknowledgements
- Conflict of InterestThe authors declare no conflict of interest.
References
- Heron, M.; Hoyert, D.L.; Murphy, S.L.; Xu, J.; Kochanek, K.D.; Tejada-Vera, B. Deaths: Final data for 2006. Natl. Vital Stat. Rep 2009, 57, 1–134. [Google Scholar]
- Yach, D.; Hawkes, C.; Gould, C.L.; Hofman, K.J. The global burden of chronic diseases: Overcoming impediments to prevention and control. JAMA J. Am. Med. Assoc 2004, 291, 2616–2622. [Google Scholar]
- Orens, J.B.; Shearon, T.H.; Freudenberger, R.S.; Conte, J.V.; Bhorade, S.M.; Ardehali, A. Thoracic organ transplantation in the United States, 1995–2004. Am. J. Transplant 2006, 6, 1188–1197. [Google Scholar]
- Trulock, E.P.; Christie, J.D.; Edwards, L.B.; Boucek, M.M.; Aurora, P.; Taylor, D.O.; Dobbels, F.; Rahmel, A.O.; Keck, B.M.; Hertz, M.I. Registry of the international society for heart and lung transplantation: Twenty-fourth official adult lung and heart-lung transplantation report—2007. J. Heart Lung Transplant 2007, 26, 782–795. [Google Scholar]
- O’Beirne, S.; Counihan, I.P.; Keane, M.P. Interstitial lung disease and lung transplantation. Semin. Respir. Crit. Care Med 2010, 31, 139–146. [Google Scholar]
- King, T.E., Jr; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar]
- Barnes, P.J. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc 2009, 6, 693–696. [Google Scholar]
- Oka, M.; Fagan, K.A.; Jones, P.L.; McMurtry, I.F. Therapeutic potential of RhoA/Rho kinase inhibitors in pulmonary hypertension. Br. J. Pharmacol 2008, 155, 444–454. [Google Scholar]
- Barlesi, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non small-cell lung cancer. J. Clin. Oncol 2007, 25, 4358–4364. [Google Scholar]
- Adcock, I.M.; Ito, K.; Barnes, P.J. Histone deacetylation: An important mechanism in inflammatory lung diseases. COPD Int. J. Chronic. Obstruct. Pulm. Dis 2005, 2, 445–455. [Google Scholar]
- Zakrzewicz, D.; Eickelberg, O. From arginine methylation to ADMA: A novel mechanism with therapeutic potential in chronic lung diseases. BMC Pulm. Med 2009, 9, 5. [Google Scholar]
- Debigare, R.; Cote, C.H.; Maltais, F. Ubiquitination and proteolysis in limb and respiratory muscles of patients with chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc 2010, 7, 84–90. [Google Scholar]
- Blanchet, F.; Schurter, B.T.; Acuto, O. Protein arginine methylation in lymphocyte signaling. Curr. Opin. Immunol 2006, 18, 321–328. [Google Scholar]
- Lake, A.N.; Bedford, M.T. Protein methylation and DNA repair. Mutat. Res 2007, 618, 91–101. [Google Scholar]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Hassa, P.O.; Gehrig, P.; Covic, M.; Hubscher, U.; Hottiger, M.O. Methylation of DNA polymerase beta by protein arginine methyltransferase 1 regulates its binding to proliferating cell nuclear antigen. FASEB J 2007, 21, 26–34. [Google Scholar]
- El-Andaloussi, N.; Valovka, T.; Toueille, M.; Steinacher, R.; Focke, F.; Gehrig, P.; Covic, M.; Hassa, P.O.; Schar, P.; Hubscher, U.; et al. Arginine methylation regulates DNA polymerase beta. Mol. Cell 2006, 22, 51–62. [Google Scholar]
- Cheng, D.; Cote, J.; Shaaban, S.; Bedford, M.T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell 2007, 25, 71–83. [Google Scholar]
- Boisvert, F.M.; Cote, J.; Boulanger, M.C.; Cleroux, P.; Bachand, F.; Autexier, C.; Richard, S. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J. Cell Biol 2002, 159, 957–969. [Google Scholar]
- Chen, D.; Ma, H.; Hong, H.; Koh, S.S.; Huang, S.M.; Schurter, B.T.; Aswad, D.W.; Stallcup, M.R. Regulation of transcription by a protein methyltransferase. Science 1999, 284, 2174–2177. [Google Scholar]
- Iberg, A.N.; Espejo, A.; Cheng, D.; Kim, D.; Michaud-Levesque, J.; Richard, S.; Bedford, M.T. Arginine methylation of the histone H3 tail impedes effector binding. J. Biol. Chem 2008, 283, 3006–3010. [Google Scholar]
- Infantino, S.; Benz, B.; Waldmann, T.; Jung, M.; Schneider, R.; Reth, M. Arginine methylation of the B cell antigen receptor promotes differentiation. J. Exp. Med 2010, 207, 711–719. [Google Scholar]
- Yu, Z.; Chen, T.; Hebert, J.; Li, E.; Richard, S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol. Cell Biol 2009, 29, 2982–2996. [Google Scholar]
- Paik, W.K.; Kim, S. Enzymatic methylation of protein fractions from calf thymus nuclei. Biochem. Biophys. Res. Commun 1967, 29, 14–20. [Google Scholar]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar]
- Lin, W.J.; Gary, J.D.; Yang, M.C.; Clarke, S.; Herschman, H.R. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J. Biol. Chem 1996, 271, 15034–15044. [Google Scholar]
- Tang, J.; Gary, J.D.; Clarke, S.; Herschman, H.R. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem 1998, 273, 16935–16945. [Google Scholar]
- Frankel, A.; Yadav, N.; Lee, J.; Branscombe, T.L.; Clarke, S.; Bedford, M.T. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem 2002, 277, 3537–3543. [Google Scholar]
- Lee, J.; Sayegh, J.; Daniel, J.; Clarke, S.; Bedford, M.T. PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J. Biol. Chem 2005, 280, 32890–32896. [Google Scholar]
- Sayegh, J.; Webb, K.; Cheng, D.; Bedford, M.T.; Clarke, S.G. Regulation of protein arginine methyltransferase 8 (PRMT8) activity by its N-terminal domain. J. Biol. Chem 2007, 282, 36444–36453. [Google Scholar]
- Branscombe, T.L.; Frankel, A.; Lee, J.H.; Cook, J.R.; Yang, Z.; Pestka, S.; Clarke, S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem 2001, 276, 32971–32976. [Google Scholar]
- Pollack, B.P.; Kotenko, S.V.; He, W.; Izotova, L.S.; Barnoski, B.L.; Pestka, S. The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J. Biol. Chem 1999, 274, 31531–31542. [Google Scholar]
- Lee, J.H.; Cook, J.R.; Yang, Z.H.; Mirochnitchenko, O.; Gunderson, S.I.; Felix, A.M.; Herth, N.; Hoffmann, R.; Pestka, S. PRMT7, a new protein arginine methyltransferase that synthesizes symmetric dimethylarginine. J. Biol. Chem 2005, 280, 3656–3664. [Google Scholar]
- Miranda, T.B.; Miranda, M.; Frankel, A.; Clarke, S. PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity. J. Biol. Chem 2004, 279, 22902–22907. [Google Scholar]
- Yildirim, A.O.; Bulau, P.; Zakrzewicz, D.; Kitowska, K.E.; Weissmann, N.; Grimminger, F.; Morty, R.E.; Eickelberg, O. Increased protein arginine methylation in chronic hypoxia: Role of protein arginine methyltransferases. Am. J. Respir. Cell Mol. Biol 2006, 35, 436–443. [Google Scholar]
- Lakowski, T.M.; Frankel, A. Kinetic analysis of human protein arginine N-methyltransferase 2: Formation of monomethyl- and asymmetric dimethyl-arginine residues on histone H4. Biochem. J 2009, 421, 253–261. [Google Scholar]
- Teerlink, T. ADMA metabolism and clearance. Vasc. Med 2005, 10, S73–S81. [Google Scholar]
- Shirakawa, T.; Kako, K.; Shimada, T.; Nagashima, Y.; Nakamura, A.; Ishida, J.; Fukamizu, A. Production of free methylarginines via the proteasome and autophagy pathways in cultured cells. Mol. Med. Report 2011, 4, 615–620. [Google Scholar]
- Bulau, P.; Zakrzewicz, D.; Kitowska, K.; Wardega, B.; Kreuder, J.; Eickelberg, O. Quantitative assessment of arginine methylation in free versus protein-incorporated amino acids in vitro and in vivo using protein hydrolysis and high-performance liquid chromatography. Biotechniques 2006, 40, 305–310. [Google Scholar]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1023–1030. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Dimethylarginine:pyruvate aminotransferase in rats. Purification, properties, and identity with alanine:glyoxylate aminotransferase 2. J. Biol. Chem 1990, 265, 20938–20945. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Watanabe, H.; Sasaoka, K. Metabolism of NG,NG-and NG,N′G-dimethylarginine in rats. Arch. Biochem. Biophys 1987, 252, 526–537. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin 2009, 59, 225–249. [Google Scholar]
- Ramalingam, S.S.; Owonikoko, T.K.; Khuri, F.R. Lung cancer: New biological insights and recent therapeutic advances. CA Cancer J. Clin 2011, 61, 91–112. [Google Scholar]
- Risch, A.; Plass, C. Lung cancer epigenetics and genetics. Int. J. Cancer 2008, 123, 1–7. [Google Scholar]
- Goulet, I.; Gauvin, G.; Boisvenue, S.; Cote, J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J. Biol. Chem 2007, 282, 33009–33021. [Google Scholar]
- Mathioudaki, K.; Papadokostopoulou, A.; Scorilas, A.; Xynopoulos, D.; Agnanti, N.; Talieri, M. The PRMT1 gene expression pattern in colon cancer. Br. J. Cancer 2008, 99, 2094–2099. [Google Scholar]
- Yoshimatsu, M.; Toyokawa, G.; Hayami, S.; Unoki, M.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Maehara, Y.; Ponder, B.A.; et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 2011, 128, 562–573. [Google Scholar]
- Smith, C.L.; Anthony, S.; Hubank, M.; Leiper, J.M.; Vallance, P. Effects of ADMA upon gene expression: An insight into the pathophysiological significance of raised plasma ADMA. PLoS Med 2005, 2, e264. [Google Scholar]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar]
- Harrison, M.J.; Tang, Y.H.; Dowhan, D.H. Protein arginine methyltransferase 6 regulates multiple aspects of gene expression. Nucleic Acids Res 2010, 38, 2201–2216. [Google Scholar] [Green Version]
- Michaud-Levesque, J.; Richard, S. Thrombospondin-1 is a transcriptional repression target of PRMT6. J. Biol. Chem 2009, 284, 21338–21346. [Google Scholar]
- Dowhan, D.H.; Harrison, M.J.; Eriksson, N.A.; Bailey, P.; Pearen, M.A.; Fuller, P.J.; Funder, J.W.; Simpson, E.R.; Leedman, P.J.; Tilley, W.D.; et al. Protein arginine methyltransferase 6-dependent gene expression and splicing: Association with breast cancer outcomes. Endocr. Relat. Cancer 2012, 19, 509–526. [Google Scholar]
- Zhong, J.; Cao, R.X.; Zu, X.Y.; Hong, T.; Yang, J.; Liu, L.; Xiao, X.H.; Ding, W.J.; Zhao, Q.; Liu, J.H.; et al. Identification and characterization of novel spliced variants of PRMT2 in breast carcinoma. FEBS J 2012, 279, 316–335. [Google Scholar]
- El Messaoudi, S.; Fabbrizio, E.; Rodriguez, C.; Chuchana, P.; Fauquier, L.; Cheng, D.; Theillet, C.; Vandel, L.; Bedford, M.T.; Sardet, C. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc. Natl. Acad. Sci. USA 2006, 103, 13351–13356. [Google Scholar]
- Majumder, S.; Liu, Y.; Ford, O.H., III; Mohler, J.L.; Whang, Y.E. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate 2006, 66, 1292–1301. [Google Scholar]
- Frietze, S.; Lupien, M.; Silver, P.A.; Brown, M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res 2008, 68, 301–306. [Google Scholar]
- Hong, H.; Kao, C.; Jeng, M.H.; Eble, J.N.; Koch, M.O.; Gardner, T.A.; Zhang, S.; Li, L.; Pan, C.X.; Hu, Z.; et al. Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer 2004, 101, 83–89. [Google Scholar]
- Ou, C.Y.; LaBonte, M.J.; Manegold, P.C.; So, A.Y.; Ianculescu, I.; Gerke, D.S.; Yamamoto, K.R.; Ladner, R.D.; Kahn, M.; Kim, J.H.; et al. A coactivator role of CARM1 in the dysregulation of β-catenin activity in colorectal cancer cell growth and gene expression. Mol. Cancer Res 2011, 9, 660–670. [Google Scholar]
- Ohira, T.; Gemmill, R.M.; Ferguson, K.; Kusy, S.; Roche, J.; Brambilla, E.; Zeng, C.; Baron, A.; Bemis, L.; Erickson, P.; et al. WNT7a induces E-cadherin in lung cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 10429–10434. [Google Scholar]
- Yue, W.; Sun, Q.; Dacic, S.; Landreneau, R.J.; Siegfried, J.M.; Yu, J.; Zhang, L. Downregulation of Dkk3 activates β-catenin/TCF-4 signaling in lung cancer. Carcinogenesis 2008, 29, 84–92. [Google Scholar]
- O’Brien, K.B.; Alberich-Jorda, M.; Yadav, N.; Kocher, O.; Diruscio, A.; Ebralidze, A.; Levantini, E.; Sng, N.J.; Bhasin, M.; Caron, T.; et al. CARM1 is required for proper control of proliferation and differentiation of pulmonary epithelial cells. Development 2010, 137, 2147–2156. [Google Scholar]
- Bulau, P.; Zakrzewicz, D.; Kitowska, K.; Leiper, J.; Gunther, A.; Grimminger, F.; Eickelberg, O. Analysis of methylarginine metabolism in the cardiovascular system identifies the lung as a major source of ADMA. Am. J. Physiol. Lung Cell Mol. Physiol 2007, 292, L18–L24. [Google Scholar]
- Hong, E.; Lim, Y.; Lee, E.; Oh, M.; Kwon, D. Tissue-specific and age-dependent expression of protein arginine methyltransferases (PRMTs) in male rat tissues. Biogerontology 2012, 13, 329–336. [Google Scholar]
- Cho, E.C.; Zheng, S.; Munro, S.; Liu, G.; Carr, S.M.; Moehlenbrink, J.; Lu, Y.C.; Stimson, L.; Khan, O.; Konietzny, R.; et al. Arginine methylation controls growth regulation by E2F-1. EMBO J 2012, 31, 1785–1797. [Google Scholar]
- Lim, J.H.; Choi, Y.J.; Cho, C.H.; Park, J.W. Protein arginine methyltransferase 5 is an essential component of the hypoxia-inducible factor 1 signaling pathway. Biochem. Biophys. Res. Commun 2012, 418, 254–259. [Google Scholar]
- Powers, M.A.; Fay, M.M.; Factor, R.E.; Welm, A.L.; Ullman, K.S. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res 2011, 71, 5579–5587. [Google Scholar]
- Jansson, M.; Durant, S.T.; Cho, E.C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol 2008, 10, 1431–1439. [Google Scholar]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res 2000, 77, 81–137. [Google Scholar]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol 2009, 49, 223–241. [Google Scholar]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar]
- Breuer, R.H.; Postmus, P.E.; Smit, E.F. Molecular pathology of non-small-cell lung cancer. Respiration 2005, 72, 313–330. [Google Scholar]
- Martinez, F.J.; Safrin, S.; Weycker, D.; Starko, K.M.; Bradford, W.Z.; King, T.E., Jr; Flaherty, K.R.; Schwartz, D.A.; Noble, P.W.; Raghu, G.; et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann. Intern. Med. 2005, 142, 963–967. [Google Scholar]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Lynch, J.P., III; Martinez, F.J. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar]
- Du Bois, R.M. Strategies for treating idiopathic pulmonary fibrosis. Nat. Rev. Drug Discov 2010, 9, 129–140. [Google Scholar]
- Li, X.; Rayford, H.; Uhal, B.D. Essential roles for angiotensin receptor AT1a in bleomycin-induced apoptosis and lung fibrosis in mice. Am. J. Pathol 2003, 163, 2523–2530. [Google Scholar]
- Konigshoff, M.; Wilhelm, A.; Jahn, A.; Sedding, D.; Amarie, O.V.; Eul, B.; Seeger, W.; Fink, L.; Gunther, A.; Eickelberg, O.; et al. The angiotensin II receptor 2 is expressed and mediates angiotensin II signaling in lung fibrosis. Am. J. Respir. Cell Mol. Biol 2007, 37, 640–650. [Google Scholar]
- Wygrecka, M.; Zakrzewicz, D.; Taborski, B.; Didiasova, M.; Kwapiszewska, G.; Preissner, K.T.; Markart, P. TGF-β1 induces tissue factor expression in human lung fibroblasts in a PI3K/JNK/Akt- and AP-1-dependent manner. Am. J. Respir. Cell Mol. Biol 2012. [Google Scholar] [CrossRef]
- Scotton, C.J.; Krupiczojc, M.A.; Konigshoff, M.; Mercer, P.F.; Lee, Y.C.; Kaminski, N.; Morser, J.; Post, J.M.; Maher, T.M.; Nicholson, A.G.; et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J. Clin. Invest 2009, 119, 2550–2563. [Google Scholar]
- Jablonska, E.; Markart, P.; Zakrzewicz, D.; Preissner, K.T.; Wygrecka, M. Transforming growth factor-β1 induces expression of human coagulation factor XII via Smad3 and JNK signaling pathways in human lung fibroblasts. J. Biol. Chem 2010, 285, 11638–11651. [Google Scholar]
- Wang, R.; Ramos, C.; Joshi, I.; Zagariya, A.; Pardo, A.; Selman, M.; Uhal, B.D. Human lung myofibroblast-derived inducers of alveolar epithelial apoptosis identified as angiotensin peptides. Am. J. Physiol 1999, 277, L1158–L1164. [Google Scholar]
- Papp, M.; Li, X.; Zhuang, J.; Wang, R.; Uhal, B.D. Angiotensin receptor subtype AT(1) mediates alveolar epithelial cell apoptosis in response to ANG II. Am. J. Physiol. Lung Cell Mol. Physiol 2002, 282, L713–L718. [Google Scholar]
- Lee, Y.H.; Mungunsukh, O.; Tutino, R.L.; Marquez, A.P.; Day, R.M. Angiotensin-II-induced apoptosis requires regulation of nucleolin and Bcl-xL by SHP-2 in primary lung endothelial cells. J. Cell Sci 2010, 123, 1634–1643. [Google Scholar]
- Bataller, R.; Sancho-Bru, P.; Gines, P.; Brenner, D.A. Liver fibrogenesis: A new role for the renin-angiotensin system. Antioxid. Redox Signal 2005, 7, 1346–1355. [Google Scholar]
- Ding, G.; Zhang, A.; Huang, S.; Pan, X.; Zhen, G.; Chen, R.; Yang, T. ANG II induces c-Jun NH2-terminal kinase activation and proliferation of human mesangial cells via redox-sensitive transactivation of the EGFR. Am. J. Physiol. Renal. Physiol 2007, 293, F1889–F1897. [Google Scholar]
- Huang, S.; Zhang, A.; Ding, G.; Chen, R. Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am. J. Physiol. Renal. Physiol 2009, 296, F1323–F1333. [Google Scholar]
- Li, X.; Zhang, H.; Soledad-Conrad, V.; Zhuang, J.; Uhal, B.D. Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. Am. J. Physiol. Lung Cell Mol. Physiol 2003, 284, L501–L507. [Google Scholar]
- Jacobi, J.; Maas, R.; Cordasic, N.; Koch, K.; Schmieder, R.E.; Boger, R.H.; Hilgers, K.F. Role of asymmetric dimethylarginine for angiotensin II-induced target organ damage in mice. Am. J. Physiol. Heart Circ. Physiol 2008, 294, H1058–H1066. [Google Scholar]
- Chen, M.F.; Xie, X.M.; Yang, T.L.; Wang, Y.J.; Zhang, X.H.; Luo, B.L.; Li, Y.J. Role of asymmetric dimethylarginine in inflammatory reactions by angiotensin II. J. Vasc. Res 2007, 44, 391–402. [Google Scholar]
- Manoury, B.; Nenan, S.; Leclerc, O.; Guenon, I.; Boichot, E.; Planquois, J.M.; Bertrand, C.P.; Lagente, V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir. Res 2005, 6, 11. [Google Scholar]
- Psathakis, K.; Mermigkis, D.; Papatheodorou, G.; Loukides, S.; Panagou, P.; Polychronopoulos, V.; Siafakas, N.M.; Bouros, D. Exhaled markers of oxidative stress in idiopathic pulmonary fibrosis. Eur. J. Clin. Invest 2006, 36, 362–367. [Google Scholar]
- Hassa, P.O.; Covic, M.; Bedford, M.T.; Hottiger, M.O. Protein arginine methyltransferase 1 coactivates NF-κB-dependent gene expression synergistically with CARM1 and PARP1. J. Mol. Biol 2008, 377, 668–678. [Google Scholar]
- Bond, M.; Chase, A.J.; Baker, A.H.; Newby, A.C. Inhibition of transcription factor NF-κB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc. Res 2001, 50, 556–565. [Google Scholar]
- Li, J.; Lau, G.K.; Chen, L.; Dong, S.S.; Lan, H.Y.; Huang, X.R.; Li, Y.; Luk, J.M.; Yuan, Y.F.; Guan, X.Y. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-κB induced matrix metalloproteinases 2 and 9 expression. PLoS One 2011, 6, e21816. [Google Scholar]
- Bond, M.; Fabunmi, R.P.; Baker, A.H.; Newby, A.C. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: An absolute requirement for transcription factor NF-κB. FEBS Lett 1998, 435, 29–34. [Google Scholar]
- Rippe, R.A.; Schrum, L.W.; Stefanovic, B.; Solis-Herruzo, J.A.; Brenner, D.A. NF-κB inhibits expression of the α1(I) collagen gene. DNA Cell Biol 1999, 18, 751–761. [Google Scholar]
- Chang, Y.I.; Hua, W.K.; Yao, C.L.; Hwang, S.M.; Hung, Y.C.; Kuan, C.J.; Leou, J.S.; Lin, W.J. Protein-arginine methyltransferase 1 suppresses megakaryocytic differentiation via modulation of the p38 MAPK pathway in K562 cells. J. Biol. Chem 2010, 285, 20595–20606. [Google Scholar]
- Kitowska, K.; Zakrzewicz, D.; Konigshoff, M.; Chrobak, I.; Grimminger, F.; Seeger, W.; Bulau, P.; Eickelberg, O. Functional role and species-specific contribution of arginases in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 2008, 294, L34–L45. [Google Scholar]
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 2008, 294, L152–L160. [Google Scholar]
- Moeller, A.; Ask, K.; Warburton, D.; Gauldie, J.; Kolb, M. The bleomycin animal model: A useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol 2008, 40, 362–382. [Google Scholar]
- Guccione, E.; Bassi, C.; Casadio, F.; Martinato, F.; Cesaroni, M.; Schuchlautz, H.; Luscher, B.; Amati, B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007, 449, 933–937. [Google Scholar]
- Chen, H.; Herndon, M.E.; Lawler, J. The cell biology of thrombospondin-1. Matrix Biol 2000, 19, 597–614. [Google Scholar]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar]
- Ghofrani, H.A.; Wilkins, M.W.; Rich, S. Uncertainties in the diagnosis and treatment of pulmonary arterial hypertension. Circulation 2008, 118, 1195–1201. [Google Scholar]
- Puri, A.; McGoon, M.D.; Kushwaha, S.S. Pulmonary arterial hypertension: Current therapeutic strategies. Nat. Clin. Pract. Cardiovasc. Med 2007, 4, 319–329. [Google Scholar]
- Ghofrani, H.A.; Barst, R.J.; Benza, R.L.; Champion, H.C.; Fagan, K.A.; Grimminger, F.; Humbert, M.; Simonneau, G.; Stewart, D.J.; Ventura, C.; et al. Future perspectives for the treatment of pulmonary arterial hypertension. J. Am. Coll. Cardiol 2009, 54, S108–S117. [Google Scholar]
- Rubin, L.J. Pulmonary arterial hypertension. Proc. Am. Thorac. Soc 2006, 3, 111–115. [Google Scholar]
- Dweik, R.A. The lung in the balance: Arginine, methylated arginines, and nitric oxide. Am. J. Physiol. Lung Cell Mol. Physiol 2007, 292, L15–L17. [Google Scholar]
- Gorenflo, M.; Zheng, C.; Werle, E.; Fiehn, W.; Ulmer, H.E. Plasma levels of asymmetrical dimethyl-L-arginine in patients with congenital heart disease and pulmonary hypertension. J. Cardiovasc. Pharmacol 2001, 37, 489–492. [Google Scholar]
- Pullamsetti, S.; Kiss, L.; Ghofrani, H.A.; Voswinckel, R.; Haredza, P.; Klepetko, W.; Aigner, C.; Fink, L.; Muyal, J.P.; Weissmann, N.; et al. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J 2005, 19, 1175–1177. [Google Scholar]
- Kielstein, J.T.; Bode-Boger, S.M.; Hesse, G.; Martens-Lobenhoffer, J.; Takacs, A.; Fliser, D.; Hoeper, M.M. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol 2005, 25, 1414–1418. [Google Scholar]
- Skoro-Sajer, N.; Mittermayer, F.; Panzenboeck, A.; Bonderman, D.; Sadushi, R.; Hitsch, R.; Jakowitsch, J.; Klepetko, W.; Kneussl, M.P.; Wolzt, M.; et al. Asymmetric dimethylarginine is increased in chronic thromboembolic pulmonary hypertension. Am. J. Respir. Crit. Care Med 2007, 176, 1154–1160. [Google Scholar]
- Landburg, P.P.; Teerlink, T.; van Beers, E.J.; Muskiet, F.A.; Kappers-Klunne, M.C.; van Esser, J.W.; Mac Gillavry, M.R.; Biemond, B.J.; Brandjes, D.P.; Duits, A.J.; et al. Association of asymmetric dimethylarginine with sickle cell disease-related pulmonary hypertension. Haematologica 2008, 93, 1410–1412. [Google Scholar]
- Dimitroulas, T.; Giannakoulas, G.; Sfetsios, T.; Karvounis, H.; Dimitroula, H.; Koliakos, G.; Settas, L. Asymmetrical dimethylarginine in systemic sclerosis-related pulmonary arterial hypertension. Rheumatology (Oxford UK) 2008, 47, 1682–1685. [Google Scholar]
- Sasaki, A.; Doi, S.; Mizutani, S.; Azuma, H. Roles of accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, and attenuated nitric oxide synthase activity in endothelial cells for pulmonary hypertension in rats. Am. J. Physiol. Lung Cell Mol. Physiol 2007, 292, L1480–L1487. [Google Scholar]
- Millatt, L.J.; Whitley, G.S.; Li, D.; Leiper, J.M.; Siragy, H.M.; Carey, R.M.; Johns, R.A. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia-induced pulmonary hypertension. Circulation 2003, 108, 1493–1498. [Google Scholar]
- Arrigoni, F.I.; Vallance, P.; Haworth, S.G.; Leiper, J.M. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation 2003, 107, 1195–1201. [Google Scholar]
- Zakrzewicz, D. Asymmetric dimethylarginine metabolism and its involvement in the pathogenesis of pulmonary arterial hypertension. Ph.D. Thesis, Justus-Liebig-Universität Gießen, Giessen, Germany, September 2008. [Google Scholar]
- Stockley, R.A.; Mannino, D.; Barnes, P.J. Burden and pathogenesis of chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc 2009, 6, 524–526. [Google Scholar]
- Stockley, R.A. Progression of chronic obstructive pulmonary disease: Impact of inflammation, comorbidities and therapeutic intervention. Curr. Med. Res. Opin 2009, 25, 1235–1245. [Google Scholar]
- Holgate, S.T. Pathogenesis of asthma. Clin. Exp. Allergy 2008, 38, 872–897. [Google Scholar]
- Parry, R.V.; Ward, S.G. Protein arginine methylation: a new handle on T lymphocytes? Trends Immunol 2010, 31, 164–169. [Google Scholar]
- Mowen, K.A.; Schurter, B.T.; Fathman, J.W.; David, M.; Glimcher, L.H. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes. Mol. Cell 2004, 15, 559–571. [Google Scholar]
- Mowen, K.A.; Tang, J.; Zhu, W.; Schurter, B.T.; Shuai, K.; Herschman, H.R.; David, M. Arginine methylation of STAT1 modulates IFNα/β-induced transcription. Cell 2001, 104, 731–741. [Google Scholar]
- Zakrzewicz, D.; Zakrzewicz, A.; Wilker, S.; Boedeker, R.H.; Padberg, W.; Eickelberg, O.; Grau, V. Dimethylarginine metabolism during acute and chronic rejection of rat renal allografts. Nephrol. Dial. Transplant 2011, 26, 124–135. [Google Scholar]
- Ahmad, T.; Mabalirajan, U.; Ghosh, B.; Agrawal, A. Altered asymmetric dimethyl arginine metabolism in allergically inflamed mouse lungs. Am. J. Respir. Cell Mol. Biol 2010, 42, 3–8. [Google Scholar]
- Scott, J.A.; North, M.L.; Rafii, M.; Huang, H.; Pencharz, P.; Subbarao, P.; Belik, J.; Grasemann, H. Asymmetric dimethylarginine is increased in asthma. Am. J. Respir. Crit. Care Med 2011, 184, 779–785. [Google Scholar]
- Klein, E.; Weigel, J.; Buford, M.C.; Holian, A.; Wells, S.M. Asymmetric dimethylarginine potentiates lung inflammation in a mouse model of allergic asthma. Am. J. Physiol. Lung Cell Mol. Physiol 2010, 299, L816–L825. [Google Scholar]
- Sun, Q.; Yang, X.; Zhong, B.; Jiao, F.; Li, C.; Li, D.; Lan, X.; Sun, J.; Lu, S. Upregulated protein arginine methyltransferase 1 by IL-4 increases eotaxin-1 expression in airway epithelial cells and participates in antigen-induced pulmonary inflammation in rats. J. Immunol 2012, 188, 3506–3512. [Google Scholar]
- Eid, H.M.; Arnesen, H.; Hjerkinn, E.M.; Lyberg, T.; Seljeflot, I. Relationship between obesity, smoking, and the endogenous nitric oxide synthase inhibitor, asymmetric dimethylarginine. Metabolism 2004, 53, 1574–1579. [Google Scholar]
- Maas, R.; Schulze, F.; Baumert, J.; Lowel, H.; Hamraz, K.; Schwedhelm, E.; Koenig, W.; Boger, R.H. Asymmetric dimethylarginine, smoking, and risk of coronary heart disease in apparently healthy men: Prospective analysis from the population-based monitoring of trends and determinants in cardiovascular disease/kooperative gesundheitsforschung in der region augsburg study and experimental data. Clin. Chem 2007, 53, 693–701. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zakrzewicz, D.; Zakrzewicz, A.; Preissner, K.T.; Markart, P.; Wygrecka, M. Protein Arginine Methyltransferases (PRMTs): Promising Targets for the Treatment of Pulmonary Disorders. Int. J. Mol. Sci. 2012, 13, 12383-12400. https://doi.org/10.3390/ijms131012383
Zakrzewicz D, Zakrzewicz A, Preissner KT, Markart P, Wygrecka M. Protein Arginine Methyltransferases (PRMTs): Promising Targets for the Treatment of Pulmonary Disorders. International Journal of Molecular Sciences. 2012; 13(10):12383-12400. https://doi.org/10.3390/ijms131012383
Chicago/Turabian StyleZakrzewicz, Dariusz, Anna Zakrzewicz, Klaus T. Preissner, Philipp Markart, and Malgorzata Wygrecka. 2012. "Protein Arginine Methyltransferases (PRMTs): Promising Targets for the Treatment of Pulmonary Disorders" International Journal of Molecular Sciences 13, no. 10: 12383-12400. https://doi.org/10.3390/ijms131012383
APA StyleZakrzewicz, D., Zakrzewicz, A., Preissner, K. T., Markart, P., & Wygrecka, M. (2012). Protein Arginine Methyltransferases (PRMTs): Promising Targets for the Treatment of Pulmonary Disorders. International Journal of Molecular Sciences, 13(10), 12383-12400. https://doi.org/10.3390/ijms131012383