Transcriptomic Analysis of Phenotypic Changes in Birch (Betula platyphylla) Autotetraploids

Abstract
:1. Introduction
2. Results
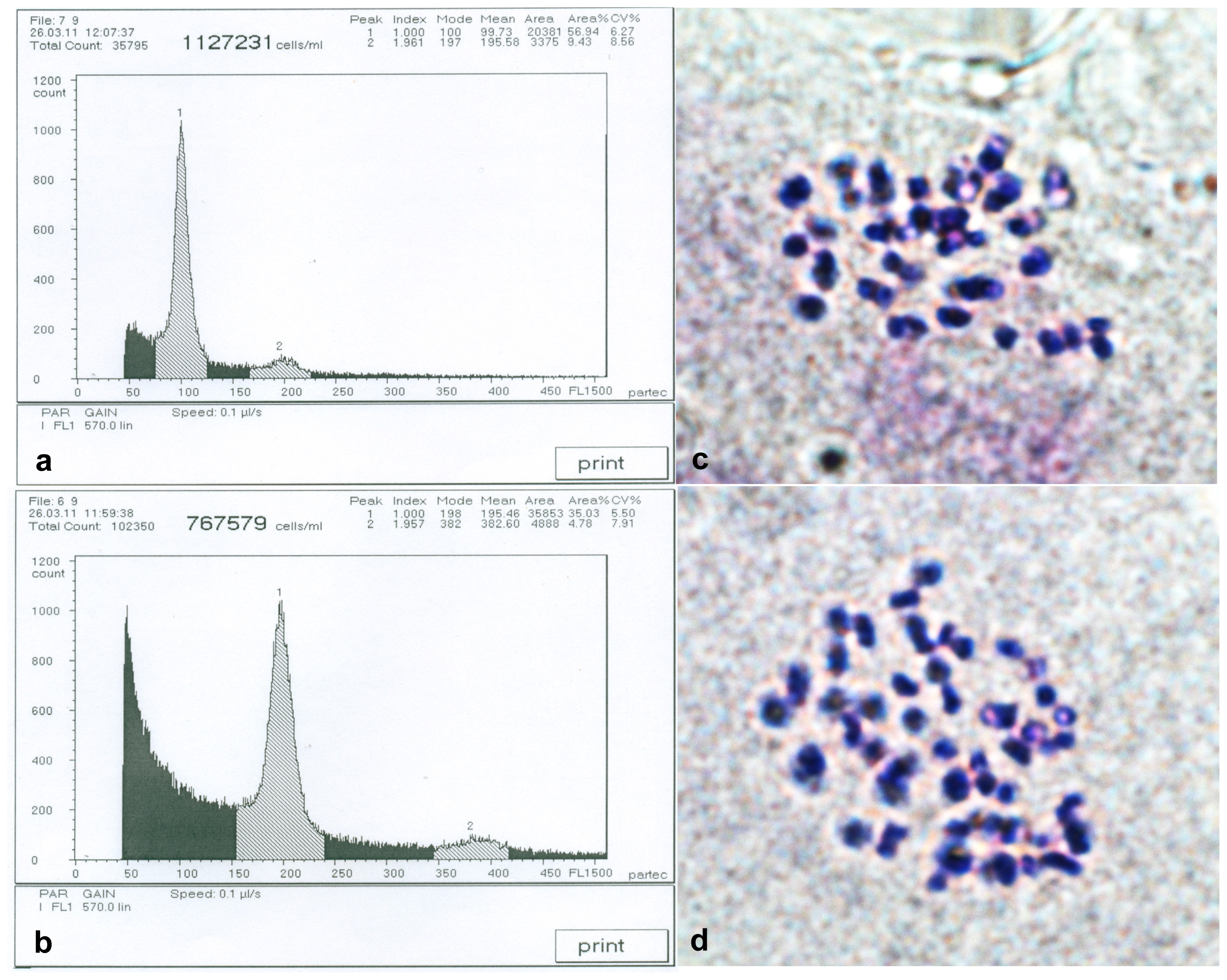
2.1. Detection of Birch Autotetraploids
2.2. Phenotypic Changes versus Ploidy in Birch
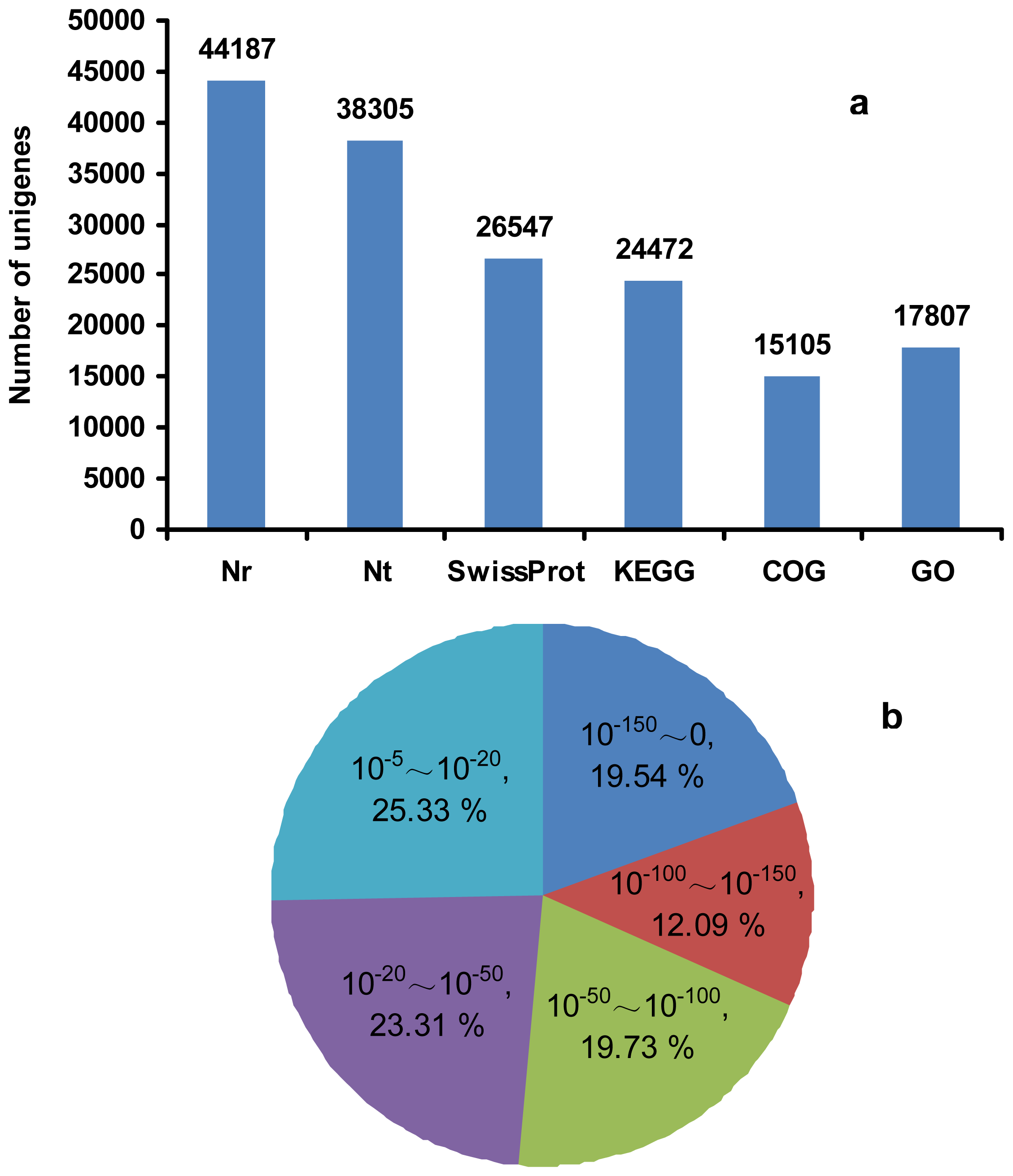
2.3. RNA-Seq, de Novo Assembly and Functional Annotation
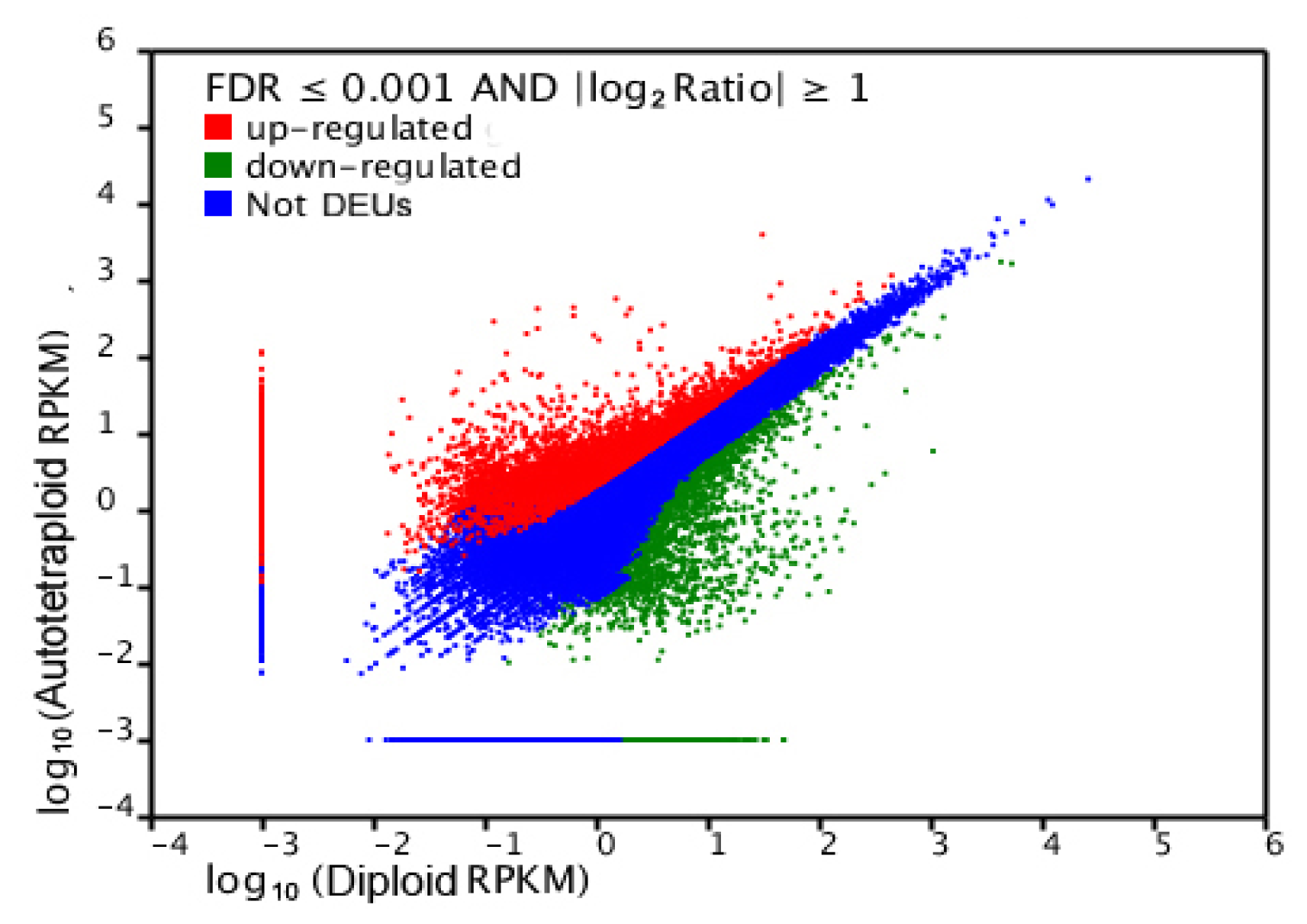
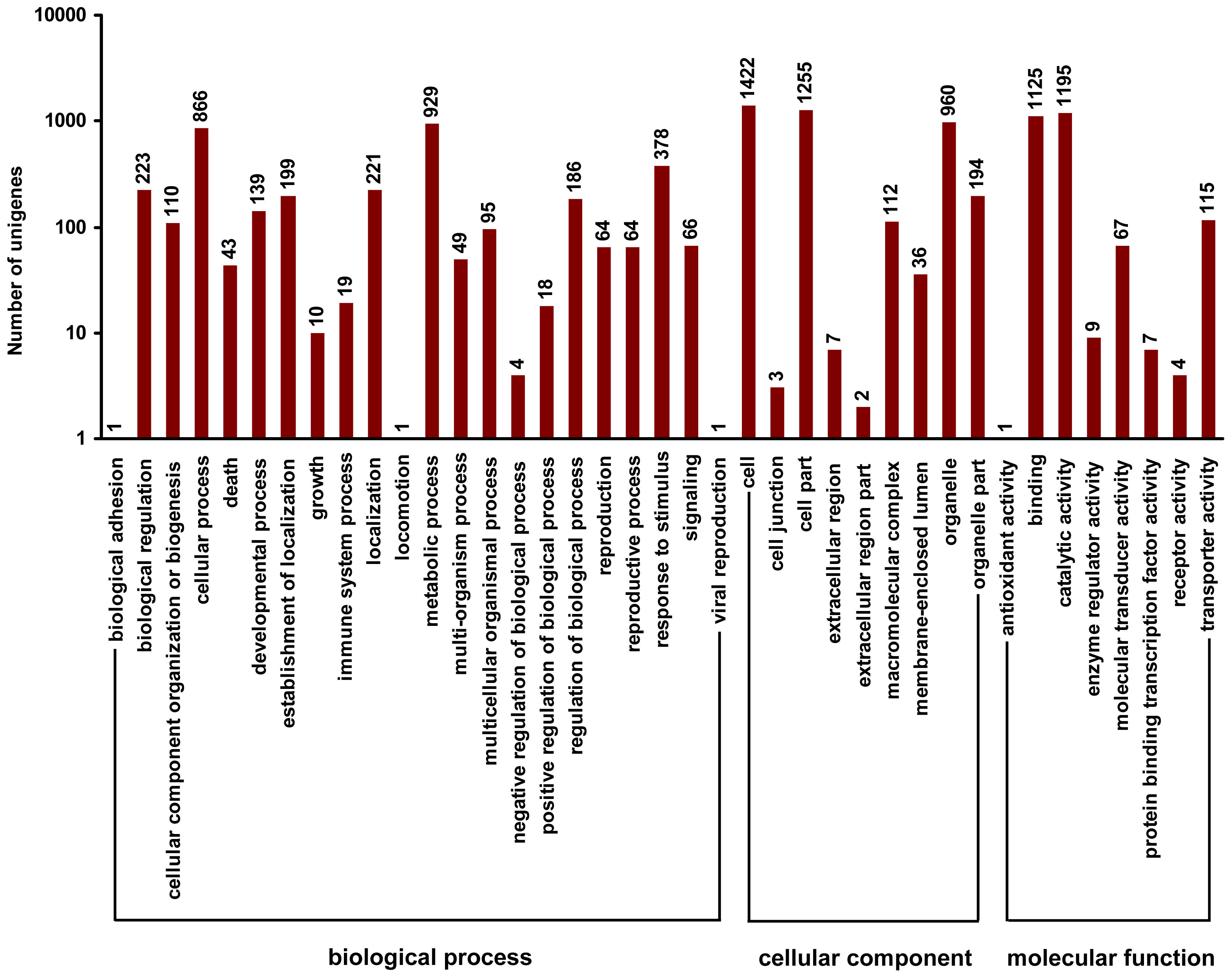
2.4. Gene Ontology Classfication of DEUs
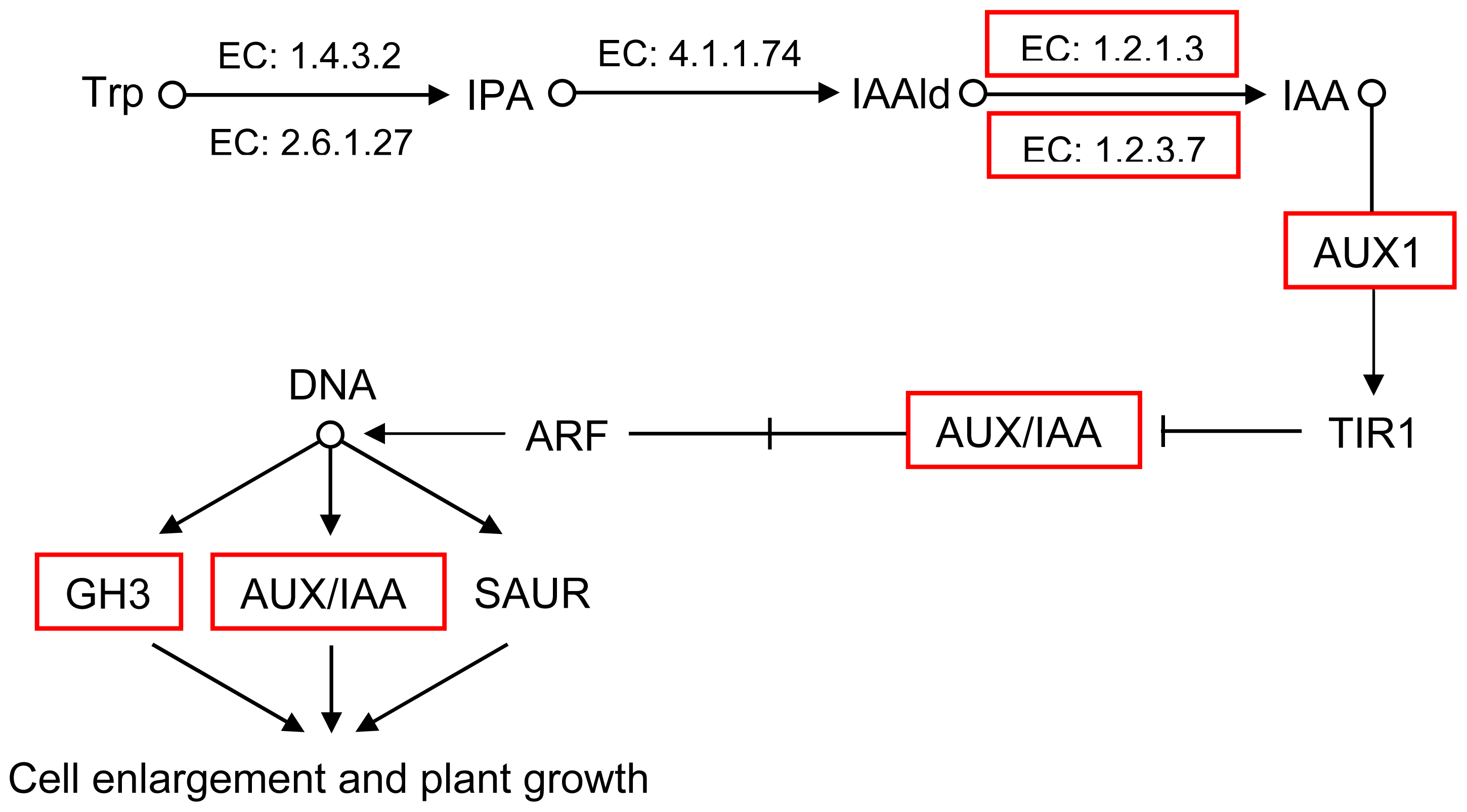
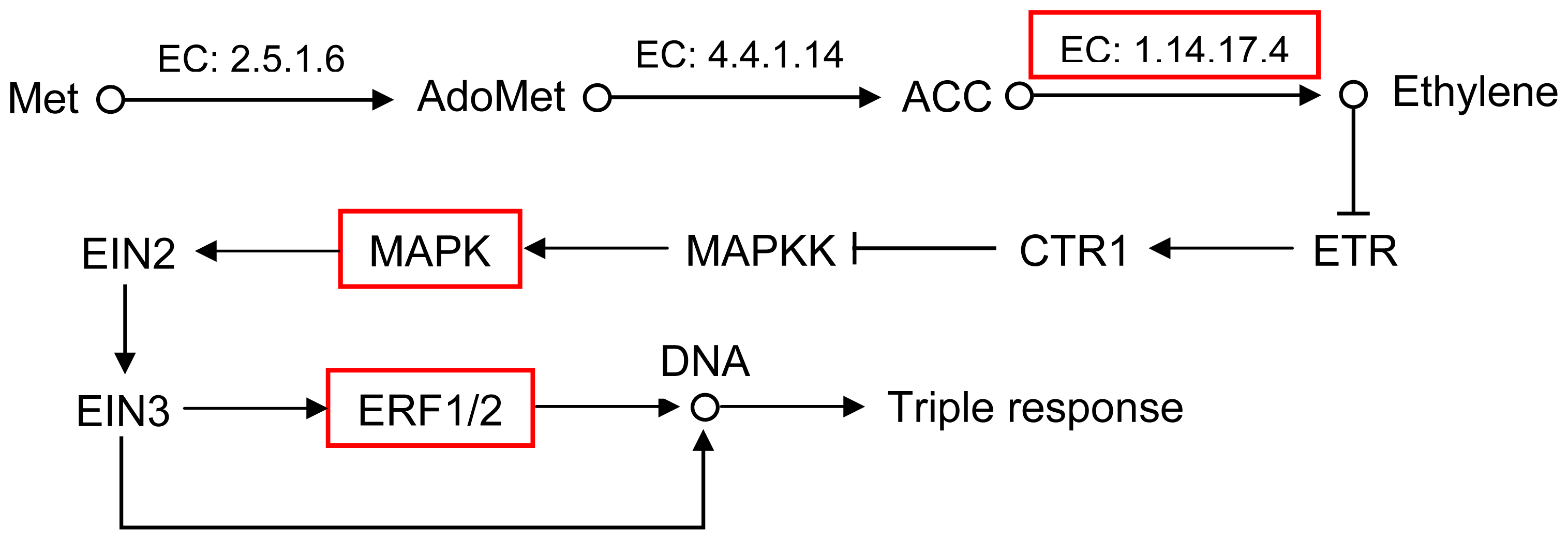
2.5. Pathway Analysis of DEUs
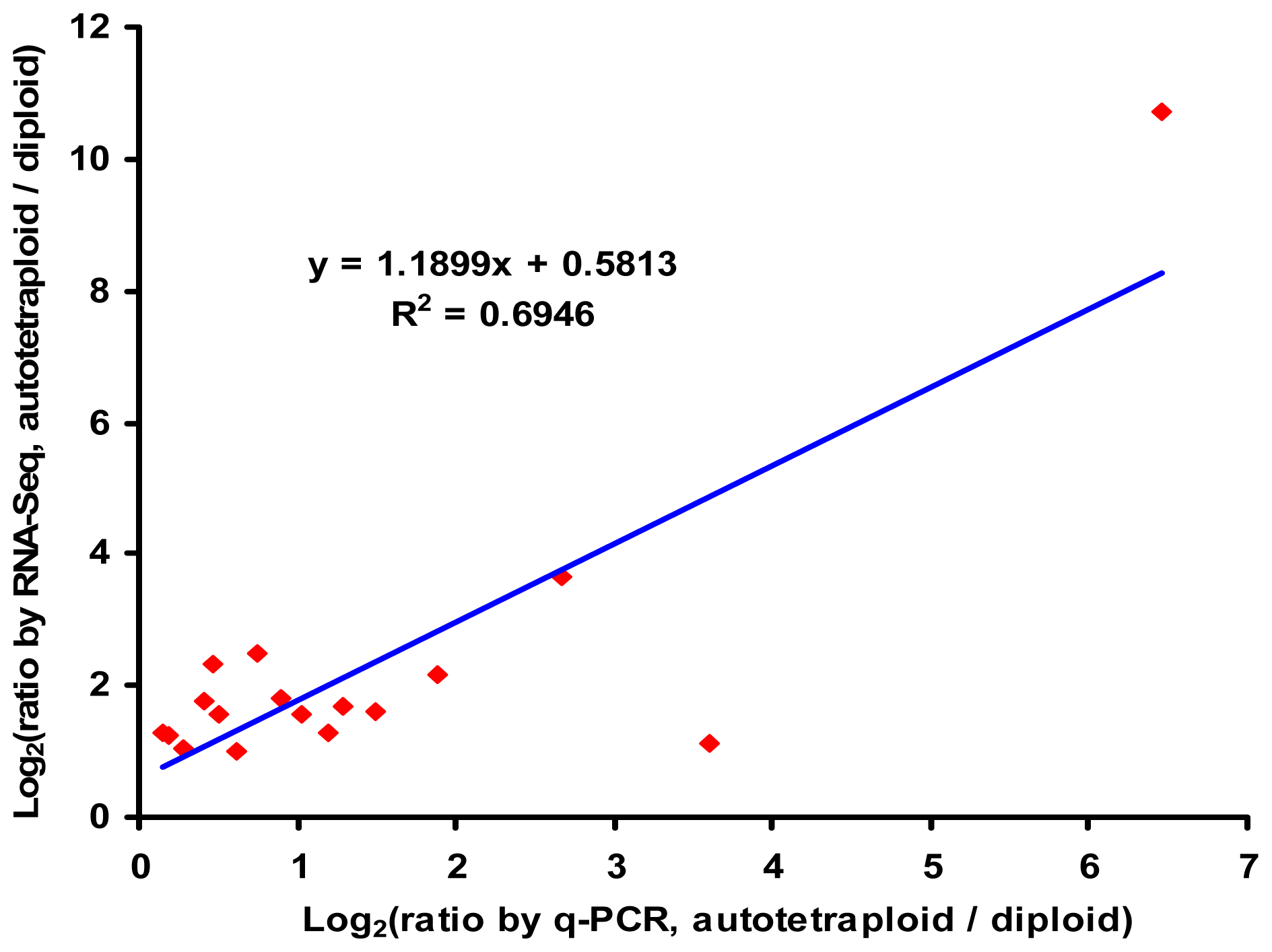
2.6. Verification of RNA-Seq by q-PCR
3. Materials and Methods
3.1. Plant Material
3.2. Ploidy Measurement
3.3. Phenotype Measurement
3.4. RNA Extraction, Library Construction and RNA-Seq
3.5. De Novo Assembly and Functional Annotation
3.6. Differential Expression of Unigenes
3.7. Gene Ontology Functional Enrichment Analysis of Differentially Expressed Unigenes (DEUs)
3.8. Pathway Analysis of DEUs
3.9. Real-Time Quantitative RT-PCR (q-PCR) Verification
4. Discussion
Acknowledgments
References
- Dwivedi, N.K.; Suryanarayana, N.; Sikdar, A.K.; Susheelamma, B.N.; Jolly, M.S. Cytomorphological studies in triploid mulberry evolved by diploidization of female gamete cells. Cytologia 1989, 54, 13–19. [Google Scholar]
- Gmitter, F.G.; Ling, X.; Deng, X. Induction of triploid Citrus plants from endosperm calli in vitro. Theor. Appl. Genet 1990, 80, 785–790. [Google Scholar]
- Leitch, A.R.; Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar]
- Einspahr, D.W.; Buijtenen, J.P.; Peckham, J.R. Natural variation and heritability in triploid aspen. Silvae Genet 1963, 12, 51–58. [Google Scholar]
- Einspahr, D.W. Production and utilization of triploid hybrid aspen. Iowa State J. Res 1984, 58, 401–409. [Google Scholar]
- Zhu, Z.; Kang, X.; Zhang, Z. Advances in the triploid breeding program of Populus tomentosa in China. J. Beijing For. Univ 1997, 6, 1–8. [Google Scholar]
- Niwa, Y.; Sasaki, Y. Plant self-defense mechanisms against oxidative injury and protection of the forest by planting trees of triploids and tetraploids. Ecotox. Environ. Safe 2003, 55, 70–81. [Google Scholar]
- Zhang, Z.; Kang, X.; Zhang, P.; Li, Y.; Wang, J. Incidence and molecular markers of 2n pollen in Populus tomentosa Carr. Euphytica 2007, 154, 145–152. [Google Scholar]
- Zhang, Z.; Kang, X. Cytological characteristics of numerically unreduced pollen production in Populus tomentosa Carr. Euphytica 2010, 173, 151–159. [Google Scholar]
- Liu, L.; Huang, F.; Luo, Q.; Pang, H.; Meng, F. cDNA-AFLP analysis of the response of tetraploid black locust (Robinia pseudoacacia L.) to salt stress. Afr. J. Biotechnol 2012, 11, 3116–3124. [Google Scholar]
- Eriksson, G.; Jonsson, A. A review of the genetics of Betula. Scand. J. For. Res 1986, 1, 421–434. [Google Scholar]
- Löve, Á. A new triploid Betula verrucosa. Svensk bot. Tidskr 1944, 38, 381–393. [Google Scholar]
- Johnsson, H. Auto- and allotriploid Betula families derived from colchicine treatment. Z. Forstgentik 1956, 5, 65–70. [Google Scholar]
- Eifler, I. The individual results of crosses between B. verrucosa and B. pubescens. Silvae Genet 1960, 9, 159–165. [Google Scholar]
- Valanne, T. Colchicine effects and colchicine-induced polyploidy in Betula. Ann. Acad. Sci. Fenn. Ser. A4 1972, 191, 1–28. [Google Scholar]
- Särkilahti, E. Micropropagation of a mature colchicine-polyploid and irradiation-mutant of Betula pendula Roth. Tree Physiol 1988, 4, 173–179. [Google Scholar]
- Pieninkeroinen, K.; Valanne, T. Old colchicines-induced polyploidy materials of Betula pendula Roth and Betula pubescens Ehrh. Ann. Sci. For 1989, 46, 264–266. [Google Scholar]
- Cameron, A.D. Autotetraploid plants from callus cultures of Betula pendula Roth. Tree Physiol 1990, 6, 229–234. [Google Scholar]
- Särkilahti, E.; Valanne, T. Induced polyploidy in Betula. Silva Fenn 1990, 24, 227–234. [Google Scholar]
- Li, W.; Berlyn, G.P.; Ashton, P.M. Polyploids and their structural and physiological characteristics relative to water deficit in Betula papyrifer. Am. J. Bot 1996, 83, 15–20. [Google Scholar]
- Stupar, R.M.; Bhaskar, P.B.; Yandell, B.S.; Rensink, W.A.; Hart, A.L.; Ouyang, S.; Veilleux, R.E.; Busse, J.S.; Erhardt, R.J.; Buell, C.R.; et al. Phenotypic and transcriptomic changes associated with potato autopolyploidization. Genetics 2007, 176, 2055–2067. [Google Scholar]
- Riddle, N.C.; Jiang, H.; An, L.; Doerge, R.W.; Birchler, J.A. Gene expression analysis at the intersection of ploidy and hybridity in maize. Theor. Appl. Genet 2010, 120, 341–353. [Google Scholar]
- Yu, Z.; Habererb, G.; Matthesa, M.; Rattei, T.; Mayer, K.F.X.; Gierl, A.; Torres-Ruiz, R.A. Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 17809–17814. [Google Scholar]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Bajgain, P.; Richardson, B.A.; Price, J.C.; Cronn, R.C.; Udall, J.A. Transcriptome characterization and polymorphism detection between subspecies of big sagebrush (Artemisia tridentata). BMC Genomics 2011, 12. [Google Scholar] [CrossRef]
- Wong, C.E.; Prem, L.B.; Harald, O.; Mohan, B.S. Transcriptional profiling of the pea shoot apical meristem reveals processes underlying its function and maintenance. BMC Plant Biol 2008, 8. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, W.; Deng, X. Relationship between ploidy variation of citrus calli and competence for somatic embryogenesis. Acta Genet. Sin 2006, 33, 647–654. [Google Scholar]
- Anamthawat-Jónsson, K. Preparation of chromosomes from plant leaf meristems for karyotype analysis and in situ hybridization. Methods Cell Sci 2003, 25, 91–95. [Google Scholar]
- Meng, X. Forest Measuration, 2nd ed; China Forestry Publishing House: Beijing, China, 2006; p. 32. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 2011, 29, 644–652. [Google Scholar]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR gene indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol 1999, 99, 138–148. [Google Scholar]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kiisttiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1666–1667. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; Kenneth, M.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res 1997, 7, 986–995. [Google Scholar]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res 2001, 125, 279–284. [Google Scholar]
- Parisod, C.; Holderegger, R.; Brochmann, C. Evolutionary consequences of autopolyploidy. New Phytol 2010, 186, 5–17. [Google Scholar]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol 2005, 8, 135–141. [Google Scholar]
- Jain, M.; Kaur, N.; Tyagi, A.K.; Khurana, J.P. The auxin-responsive GH3 gene family in rice (Oryza sativa). Funct. Integr. Genomics 2006, 6, 36–46. [Google Scholar]
- Mashiguchi, K.; Tanaka, K.; Sakai, T.; Sugawara, S.; Kawaide, H.; Natsume, M.; Hanada, A.; Yaeno, T.; Shirasu, K.; Yao, H.; et al. The main auxin biosynthesis pathway in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18512–18517. [Google Scholar]
- Mikkelsen, M.D.; Naur, P.; Halkier, B.A. Arabidopsis mutants in the C–S lyase of glucosinolate biosynthesis establish a critical role for indole-3-acetaldoxime in auxin homeostasis. Plant J 2004, 37, 770–777. [Google Scholar]
- Puyvelde, S.; Cloots, L.; Engelen, K.; Das, F.; Marchal, K.; Vanderleyden, J.; Spaepen, S. Transcriptome analysis of the rhizosphere bacterium Azospirillum brasilense reveals an extensive auxin response. Microb. Ecol 2011, 61, 723–728. [Google Scholar]
- Vandenbussche, F.; Petrášek, J.; Zádníková, P.; Hoyerová, K.; Pešek, B.; Raz, V.; Swarup, R.; Bennett, M.; Zažímalová, E.; Benková, E.; et al. The auxin influx carriers AUX1 and LAX3 are involved in auxin-ethylene interactions during apical hook development in Arabidopsis thaliana seedlings. Development 2010, 137, 597–606. [Google Scholar]
- Liscum, E.; Reed, J.W. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol 2002, 49, 387–400. [Google Scholar]
- Bleecker, A.B.; Estelle, M.A.; Somerville, C.; Kende, H. Insensitivity to ethylene conferred by a dominant mutation in Arabidopsis thaliana. Science 1988, 241, 1086–1089. [Google Scholar]
- Yang, S.; Hoffman, N.E. Ethylene biosynthesis and its regulation in higher plants. Annu. Rev. Plant Physiol 1984, 35, 155–189. [Google Scholar]
- Ouaked, F.; Rozhon, W.; Lecourieux, D.; Hirt, H. A MAPK pathway mediates ethylene signaling in plants. EMBO J 2003, 22, 1282–1288. [Google Scholar]
- Chen, Y.; Etheridge, N.; Schaller, G.E. Ethylene signal transduction. Ann. Bot-Lond 2005, 95, 901–915. [Google Scholar]
- Nakano, T.; Nishiuchi, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Studies on transcriptional regulation of endogenous genes by ERF2 transcription factor in tobacco cells. Plant Cell Physiol 2006, 47, 554–558. [Google Scholar]
- Adams, K.L. Evolution of duplicate gene expression in polyploidy and hybrid plants. J. Hered 2007, 98, 136–141. [Google Scholar]
- Nie, J.; Stewart, R.; Zhang, H.; Thomson, J.A.; Ruan, F.; Cui, X.; Wei, H. TF-Cluster: A pipeline for identifying functionally coordinated transcription factors via network decomposition of the shared coexpression connectivity matrix (SCCM). BMC Syst. Biol 2011, 5. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Full-sib families | |||||||
|---|---|---|---|---|---|---|---|---|
| 5 × 3 | 5 × 9 | 5 × 11 | 6 × 5 | |||||
| Diploid | Autotetraploid | Diploid | Autotetraploid | Diploid | Autotetraploid | Diploid | Autotetraploid | |
| Height (m) | 6.80 ± 0.25 A | 5.61 ± 0.49 B | 6.86 ± 0.17 A | 5.70 ± 0.43 B | 6.83 ± 0.22 A | 5.52 ± 0.48 B | 6.92 ± 0.18 A | 5.48 ± 0.37 B |
| Breast-height diameter (mm) | 36.44 ± 1.13 B | 46.71 ± 3.00 A | 36.59 ± 0.91 B | 47.30 ± 3.02 A | 37.88 ± 1.80 B | 46.05 ± 3.35 A | 37.59 ± 0.83 B | 45.70 ± 3.12 A |
| Volume (dm3) | 4.09 ± 0.29 B | 5.96 ± 1.07 A | 4.15 ± 0.24 B | 6.17 ± 1.02 A | 4.45 ± 0.52 B | 5.74 ± 1.02 A | 4.41 ± 0.25 B | 5.61 ± 0.91 A |
| Leaf area (cm2) | 13.92 ± 5.62 B | 28.81 ± 8.63 A | 20.77 ± 4.82 B | 26.44 ± 5.57 A | 13.51 ± 3.87 B | 30.64 ± 8.90 A | 14.48 ± 3.97 B | 27.83 ± 6.40 A |
| Fruit length (mm) | 40.70 ± 2.53 B | 48.17 ± 5.25 A | 44.51 ± 5.22 B | 50.87 ± 6.06 A | 45.62 ± 3.59 B | 48.53 ± 5.07 A | 51.78 ± 6.07 B | 57.63 ± 8.39 A |
| Fruit diameter (mm) | 8.55 ± 0.58 B | 11.43 ± 2.88 A | 8.02 ± 0.53 B | 12.76 ± 2.04 A | 9.11 ± 0.59 B | 12.03 ± 1.19 A | 9.02 ± 0.77 B | 11.31 ± 1.46 A |
| Stoma length (μm) | 11.09 ± 1.33 B | 23.19 ± 5.06 A | 16.73 ± 2.04 B | 26.31 ± 4.17 A | 11.78 ± 1.60 B | 27.28 ± 4.64 A | 11.84 ± 1.34 B | 20.81 ± 3.43 A |
| Samples | Total reads | Total nucleotides (nt) | Q20 percentage (%) | N percentage (%) | GC percentage (%) |
|---|---|---|---|---|---|
| Diploid | 51,251,240 | 4,612,611,600 | 96.69 | 0.00 | 48.53 |
| Autotetraploid | 54,936,162 | 4,944,254,580 | 96.65 | 0.00 | 48.28 |
| Samples | Number of unigenes | Total length of unigenes (nt) | Mean length of unigenes (nt) | N50 (nt) |
|---|---|---|---|---|
| Diploid | 78,213 | 48,907,808 | 625 | 1115 |
| Autotetraploid | 88,607 | 55,149,462 | 622 | 1117 |
| All | 84,788 | 64,593,413 | 762 | 1294 |
| Genes | Log2(autotetraploid RPKM/diploid RPKM) | Annotation |
|---|---|---|
| GI: 224112551 | 1.8 | auxin influx carrier component |
| GI: 302398569 | 1.6 | ARF domain class transcription factor |
| GI: 114733 | 1.6 | RecName: Full = Auxin-induced protein AUX22 |
| GI: 20269055 | 1.5 | AUX/IAA protein |
| GI: 62125392 | 1.3 | auxin-responsive protein IAA |
| GI: 302398589 | 1.3 | ARF domain class transcription factor |
| GI: 42561642 | 1.2 | P-loop containing nucleoside triphosphate hydrolase-like protein |
| GI: 50404477 | 1 | IAA type protein |
| GI: 307136360 | 1.1 | auxin-regulated protein |
| GI: 297802558 | 1.8 | ALDH3I1 |
| GI: 224131694 | 2.3 | aldehyde oxidase 2 |
| Genes | Log2(autotetraploid RPKM/diploid RPKM) | Annotation |
|---|---|---|
| GI: 115345808 | 2.5 | mitogen-activated protein kinase 20 |
| GI: 224123518 | 10.7 | AP2/ERF domain-containing transcription factor |
| GI: 292668935 | 3.7 | AP2 domain class transcription factor |
| GI: 289466349 | 1.7 | ERF transcription factor 4 |
| GI: 222427679 | 2.2 | ethylene responsive transcription factor 1a |
| GI: 62526579 | 1 | ACC oxidase ACCO2 |
| Gene-ID | Forward primer (5′-3′) | Reverse primer (5′-3′) | Product size (bp) |
|---|---|---|---|
| 18S rRNA | ATCTTGGGTTGGGCAGATCG | CATTACTCCGATCCCGAAGG | 223 |
| α-Tubulin | GCACTGGCCTCCAAGGAT | TGGGTCGCTCAATGTCAAGG | 282 |
| Unigene20020_All | AGACTGAGATTTGGGTATCTTCG | CGTTAACAGAACCTGAGCAACC | 259 |
| CL26738.Contig1_All | AGACGATGATGATGAATCTTTGC | CCTCAGAAAGATCGACCTAAGG | 298 |
| CL37007.Contig1_All | GAACTTGTTCTGAACTGTCTGC | GGAAACTTCAAACGAAGACC | 273 |
| Unigene32315_All | GTCTTTCTCCCTCCTTGGTTAGC | TTGTTCTTCCTTAATTTCGTGTCC | 273 |
| CL41914.Contig1_All | GCATTGGAAACATTAGAACTGTCC | CCTTCCTTATCGTCAAGCAATCG | 264 |
| CL10694.Contig1_All | CCAAGGAACATCTCCTGCAAGC | AGCTCCATCAGCATCAAGGTGG | 290 |
| CL1467.Contig1_All | GCTCTGGTTGGTGGAATGG | CCTCAGCTCTGTCTCCTCG | 248 |
| CL26579.Contig1_All | CCAGCTGCAAAGACACAAGTGG | TCCAACTAGCATCCAGTCACC | 294 |
| CL7645.Contig1_All | GGTTTGAGCAGTTTTGACATAGC | CATCGATGAAGGCAATTCAGTCC | 277 |
| CL20989.Contig1_All | GTTCTTGAGAGAGAGAGAGAGG | TACACTGCTTTCGTACGTGTGG | 284 |
| Unigene39140_All | TGAGCTCTCCAGTGTCGACC | GGAACCCATCTCTGCTATTTCC | 251 |
| Unigene25063_All | CGACCTACTGCAGAAGAGG | TCCTCAAGATATGCAAACTGC | 272 |
| Unigene31405_All | CCAACCTTTCTACTATTTTCTGC | GTGGTTTTGGATGAGTTGTTAGC | 244 |
| Unigene29640_All | TTGTCGAATCCCTTATCTCAGC | TCACCGGAATCTTTCTCCTGG | 265 |
| CL29218.Contig1_All | GGATGAAATCTTTGCAAGCTGG | GCACCAAATCAATGCGTCGAGC | 279 |
| CL6280.Contig1_All | TTGGAGCACGAAAGTTTACC | GAGTGGGAAGTTGAGCAAGG | 279 |
| CL27664.Contig1_All | CTTCTAGGCAGTGACACTATGG | GTGAACATAGGAGACCAAGTGG | 285 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mu, H.-Z.; Liu, Z.-J.; Lin, L.; Li, H.-Y.; Jiang, J.; Liu, G.-F. Transcriptomic Analysis of Phenotypic Changes in Birch (Betula platyphylla) Autotetraploids. Int. J. Mol. Sci. 2012, 13, 13012-13029. https://doi.org/10.3390/ijms131013012
Mu H-Z, Liu Z-J, Lin L, Li H-Y, Jiang J, Liu G-F. Transcriptomic Analysis of Phenotypic Changes in Birch (Betula platyphylla) Autotetraploids. International Journal of Molecular Sciences. 2012; 13(10):13012-13029. https://doi.org/10.3390/ijms131013012
Chicago/Turabian StyleMu, Huai-Zhi, Zi-Jia Liu, Lin Lin, Hui-Yu Li, Jing Jiang, and Gui-Feng Liu. 2012. "Transcriptomic Analysis of Phenotypic Changes in Birch (Betula platyphylla) Autotetraploids" International Journal of Molecular Sciences 13, no. 10: 13012-13029. https://doi.org/10.3390/ijms131013012
APA StyleMu, H. -Z., Liu, Z. -J., Lin, L., Li, H. -Y., Jiang, J., & Liu, G. -F. (2012). Transcriptomic Analysis of Phenotypic Changes in Birch (Betula platyphylla) Autotetraploids. International Journal of Molecular Sciences, 13(10), 13012-13029. https://doi.org/10.3390/ijms131013012