Molecular Diagnostic and Pathogenesis of Hereditary Hemochromatosis

Abstract
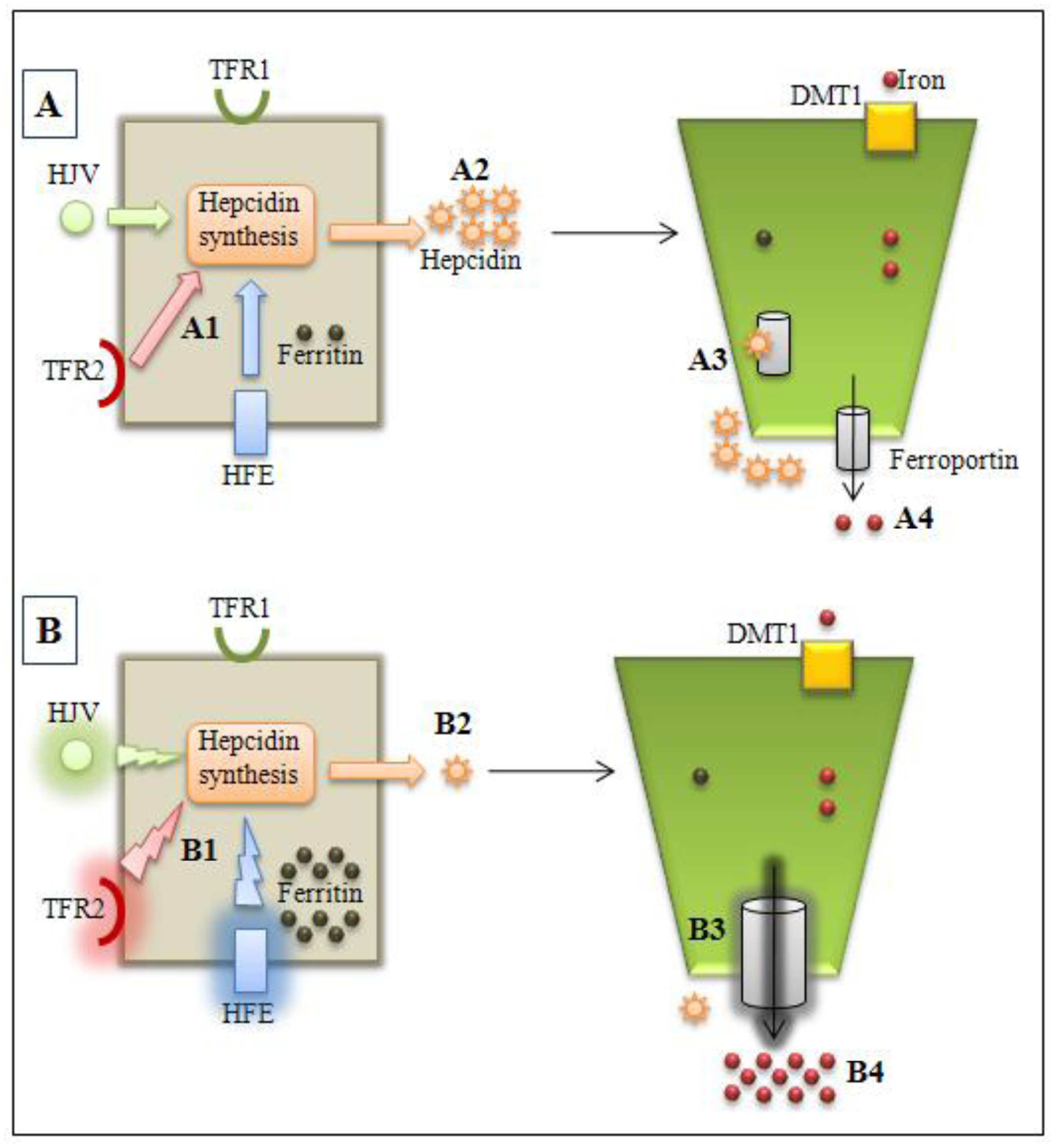
:1. Introduction
2. HH Types, Related Genes and Their Main Mutations
2.1. HFE
2.2. HJV and HAMP
2.3. TFR2
2.4. SLC40A1
3. Biochemical Assays for Body Iron Store Analysis
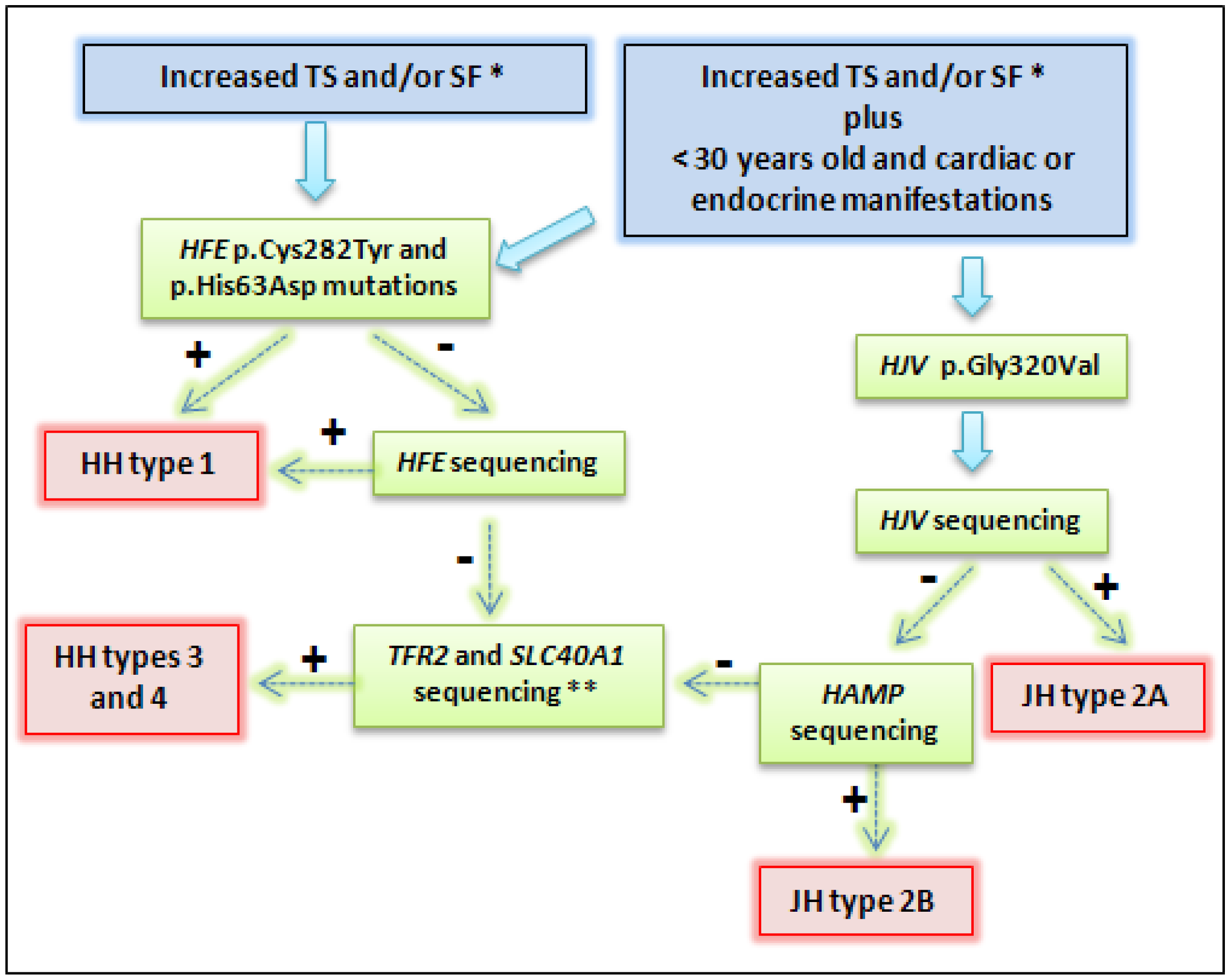
4. Genetic Testing and Methodology
4.1. Genetic Testing
4.2. Methodology
5. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Alexander, J.; Kowdley, K.V. HFE-associated hereditary hemochromatosis. Genet. Med 2009, 11, 307–313. [Google Scholar]
- Bacon, B.R. Hemochromatosis: Diagnosis and management. Gastroenterology 2001, 120, 718–725. [Google Scholar]
- Moyer, T.P.; Highsmith, W.E.; Smyrk, T.C.; Gross, J.B., Jr. Hereditary hemochromatosis: Laboratory evaluation. Clin. Chim. Acta 2011, 412, 1485–1492. [Google Scholar]
- Phatak, P.; Brissot, P.; Wurster, M.; Adams, P.C.; Bonkovsky, H.L.; Gross, J.; Malfertheiner, P.; McLaren, G.D.; Niederau, C.; Piperno, A.; et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010, 52, 1671–1779. [Google Scholar]
- Santos, P.C.; Cancado, R.D.; Pereira, A.C.; Chiattone, C.S.; Krieger, J.E.; Guerra-Shinohara, E.M. HJV hemochromatosis, iron overload, and hypogonadism in a Brazilian man: Treatment with phlebotomy and deferasirox.
- Dupradeau, F.Y.; Pissard, S.; Coulhon, M.P.; Cadet, E.; Foulon, K.; Fourcade, C.; Goossens, M.; Case, D.A.; Rochette, J. An unusual case of hemochromatosis due to a new compound heterozygosity in HFE (p.[Gly43Asp;His63Asp]+[Cys282Tyr]): Structural implications with respect to binding with transferrin receptor 1. Hum. Mutat 2008, 29. [Google Scholar] [CrossRef]
- Mendes, A.I.; Ferro, A.; Martins, R.; Picanco, I.; Gomes, S.; Cerqueira, R.; Correia, M.; Nunes, A.R.; Esteves, J.; Fleming, R.; Faustino, P. Non-classical hereditary hemochromatosis in Portugal: Novel mutations identified in iron metabolism-related genes. Ann. Hematol 2009, 88, 229–234. [Google Scholar]
- Pietrangelo, A. Molecular insights into the pathogenesis of hereditary haemochromatosis. Gut 2006, 55, 564–568. [Google Scholar]
- Swinkels, D.W.; Janssen, M.C.; Bergmans, J.; Marx, J.J. Hereditary hemochromatosis: Genetic complexity and new diagnostic approaches. Clin. Chem 2006, 52, 950–968. [Google Scholar]
- Merryweather-Clarke, A.T.; Pointon, J.J.; Shearman, J.D.; Robson, K.J. Global prevalence of putative haemochromatosis mutations. J. Med. Genet 1997, 34, 275–278. [Google Scholar]
- Nemeth, E.; Ganz, T. The role of hepcidin in iron metabolism. Acta Haematol 2009, 122, 78–86. [Google Scholar]
- Santos, P.C.; Cancado, R.D.; Terada, C.T.; Rostelato, S.; Gonzales, I.; Hirata, R.D.; Hirata, M.H.; Chiattone, C.S.; Guerra-Shinohara, E.M. HFE gene mutations and iron status of Brazilian blood donors. Braz. J. Med. Biol. Res 2010, 43, 107–114. [Google Scholar] [Green Version]
- Terada, C.T.; Santos, P.C.; Cancado, R.D.; Rostelato, S.; Lopreato, F.R.; Chiattone, C.S.; Guerra-Shinohara, E.M. Iron deficiency and frequency of HFE C282Y gene mutation in Brazilian blood donors. Transfus. Med 2009, 19, 245–251. [Google Scholar]
- Lok, C.Y.; Merryweather-Clarke, A.T.; Viprakasit, V.; Chinthammitr, Y.; Srichairatanakool, S.; Limwongse, C.; Oleesky, D.; Robins, A.J.; Hudson, J.; Wai, P.; et al. Iron overload in the Asian community. Blood 2009, 114, 20–25. [Google Scholar]
- Santos, P.C.; Cancado, R.D.; Pereira, A.C.; Schettert, I.T.; Soares, R.A.; Pagliusi, R.A.; Hirata, R.D.; Hirata, M.H.; Teixeira, A.C.; Figueiredo, M.S.; et al. Hereditary hemochromatosis: Mutations in genes involved in iron homeostasis in Brazilian patients. Blood Cells Mol. Dis 2010, 46, 302–307. [Google Scholar]
- Kaplan, J.; Ward, D.M.; de Domenico, I. The molecular basis of iron overload disorders and iron-linked anemias. Int. J. Hematol 2011, 93, 14–20. [Google Scholar]
- Lee, P.L.; Beutler, E. Regulation of hepcidin and iron-overload disease. Annu. Rev. Pathol 2009, 4, 489–515. [Google Scholar]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, e1–e2. [Google Scholar]
- Pietrangelo, A. Hepcidin in human iron disorders: Therapeutic implications. J. Hepatol 2011, 54, 173–181. [Google Scholar]
- Adams, P.C. Nonexpressing homozygotes for C282Y hemochromatosis: Minority or majority of cases? Mol. Genet. Metab 2000, 71, 81–86. [Google Scholar]
- Leone, P.E.; Gimenez, P.; Collantes, J.C.; Paz-y-Mino, C. Analysis of HFE gene mutations (C282Y, H63D, and S65C) in the Ecuadorian population. Ann. Hematol 2005, 84, 103–105. [Google Scholar]
- Potekhina, E.S.; Lavrov, A.V.; Samokhodskaya, L.M.; Efimenko, A.Y.; Balatskiy, A.V.; Baev, A.A.; Litvinova, M.M.; Nikitina, L.A.; Shipulin, G.A.; Bochkov, N.P.; et al. Unique genetic profile of hereditary hemochromatosis in Russians: High frequency of C282Y mutation in population, but not in patients. Blood Cells Mol. Dis 2005, 35, 182–188. [Google Scholar]
- Santos, P.C.; Pereira, A.C.; Cancado, R.D.; Schettert, I.T.; Sobreira, T.J.; Oliveira, P.S.; Hirata, R.D.; Hirata, M.H.; Figueiredo, M.S.; Chiattone, C.S.; et al. HFE gene mutations in patients with primary iron overload: Is there a significant improvement in molecular diagnosis yield with HFE sequencing? Blood Cells Mol. Dis 2010, 45, 302–307. [Google Scholar]
- Thakur, V.; Guptan, R.C.; Hashmi, A.Z.; Sakhuja, P.; Malhotra, V.; Sarin, S.K. Absence of hemochromatosis associated Cys282Tyr HFE gene mutation and low frequency of hemochromatosis phenotype in nonalcoholic chronic liver disease patients in India. J. Gastroenterol. Hepatol 2004, 19, 86–90. [Google Scholar]
- Pelucchi, S.; Mariani, R.; Bertola, F.; Arosio, C.; Piperno, A. Homozygous deletion of HFE: The Sardinian hemochromatosis. Blood 2009, 113. [Google Scholar] [CrossRef]
- Bennett, M.J.; Lebron, J.A.; Bjorkman, P.J. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature 2000, 403, 46–53. [Google Scholar]
- Ahmad, K.A.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Britton, R.S.; Bacon, B.R.; Sly, W.S.; Fleming, R.E. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol. Dis 2002, 29, 361–366. [Google Scholar]
- Bridle, K.R.; Frazer, D.M.; Wilkins, S.J.; Dixon, J.L.; Purdie, D.M.; Crawford, D.H.; Subramaniam, V.N.; Powell, L.W.; Anderson, G.J.; Ramm, G.A. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 2003, 361, 669–673. [Google Scholar]
- Gehrke, S.G.; Pietrangelo, A.; Kascak, M.; Braner, A.; Eisold, M.; Kulaksiz, H.; Herrmann, T.; Hebling, U.; Bents, K.; Gugler, R.; et al. HJV gene mutations in European patients with juvenile hemochromatosis. Clin. Genet 2005, 67, 425–428. [Google Scholar]
- Corradini, E.; Garuti, C.; Montosi, G.; Ventura, P.; Andriopoulos, B., Jr; Lin, H.Y.; Pietrangelo, A.; Babitt, J.L. Bone morphogenetic protein signaling is impaired in an HFE knockout mouse model of hemochromatosis. Gastroenterology 2009, 137, 1489–1497. [Google Scholar]
- Kautz, L.; Meynard, D.; Besson-Fournier, C.; Darnaud, V.; Al Saati, T.; Coppin, H.; Roth, M.P. BMP/Smad signaling is not enhanced in Hfe-deficient mice despite increased Bmp6 expression. Blood 2009, 114, 2515–2520. [Google Scholar]
- Papanikolaou, G.; Samuels, M.E.; Ludwig, E.H.; MacDonald, M.L.; Franchini, P.L.; Dube, M.P.; Andres, L.; MacFarlane, J.; Sakellaropoulos, N.; Politou, M.; et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet 2004, 36, 77–82. [Google Scholar]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet 2003, 33, 21–22. [Google Scholar]
- Lin, L.; Goldberg, Y.P.; Ganz, T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood 2005, 106, 2884–2889. [Google Scholar]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar]
- Aguilar-Martinez, P.; Lok, C.Y.; Cunat, S.; Cadet, E.; Robson, K.; Rochette, J. Juvenile hemochromatosis caused by a novel combination of hemojuvelin G320V/R176C mutations in a 5-year old girl. Haematologica 2007, 92, 421–422. [Google Scholar]
- De Lima Santos, P.C.; Pereira, A.C.; Cancado, R.D.; Schettert, I.T.; Hirata, R.D.; Hirata, M.H.; Figueiredo, M.S.; Chiattone, C.S.; Krieger, J.E.; Guerra-Shinohara, E.M. Hemojuvelin and hepcidin genes sequencing in Brazilian patients with primary iron overload. Genet. Test Mol. Biomark 2010, 14, 803–806. [Google Scholar]
- Lanzara, C.; Roetto, A.; Daraio, F.; Rivard, S.; Ficarella, R.; Simard, H.; Cox, T.M.; Cazzola, M.; Piperno, A.; Gimenez-Roqueplo, A.P.; et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood 2004, 103, 4317–4321. [Google Scholar]
- Lee, P.L.; Beutler, E.; Rao, S.V.; Barton, J.C. Genetic abnormalities and juvenile hemochromatosis: Mutations of the HJV gene encoding hemojuvelin. Blood 2004, 103, 4669–4671. [Google Scholar]
- Jacolot, S.; Le Gac, G.; Scotet, V.; Quere, I.; Mura, C.; Ferec, C. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood 2004, 103, 2835–2840. [Google Scholar]
- Porto, G.; Roetto, A.; Daraio, F.; Pinto, J.P.; Almeida, S.; Bacelar, C.; Nemeth, E.; Ganz, T.; Camaschella, C. A Portuguese patient homozygous for the -25G>A mutation of the HAMP promoter shows evidence of steady-state transcription but fails to up-regulate hepcidin levels by iron. Blood 2005, 106, 2922–2923. [Google Scholar]
- Biasiotto, G.; Roetto, A.; Daraio, F.; Polotti, A.; Gerardi, G.M.; Girelli, D.; Cremonesi, L.; Arosio, P.; Camaschella, C. Identification of new mutations of hepcidin and hemojuvelin in patients with HFE C282Y allele. Blood Cells Mol. Dis 2004, 33, 338–343. [Google Scholar]
- Altes, A.; Bach, V.; Ruiz, A.; Esteve, A.; Felez, J.; Remacha, A.F.; Sarda, M.P.; Baiget, M. Mutations in HAMP and HJV genes and their impact on expression of clinical hemochromatosis in a cohort of 100 Spanish patients homozygous for the C282Y mutation of HFE gene. Ann. Hematol 2009, 88, 951–955. [Google Scholar]
- Merryweather-Clarke, A.T.; Cadet, E.; Bomford, A.; Capron, D.; Viprakasit, V.; Miller, A.; McHugh, P.J.; Chapman, R.W.; Pointon, J.J.; Wimhurst, V.L.; et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet 2003, 12, 2241–2247. [Google Scholar]
- Camaschella, C.; Poggiali, E. Rare types of genetic hemochromatosis. Acta Haematol 2009, 122, 140–145. [Google Scholar]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet 2006, 38, 531–539. [Google Scholar]
- Papanikolaou, G.; Tzilianos, M.; Christakis, J.I.; Bogdanos, D.; Tsimirika, K.; MacFarlane, J.; Goldberg, Y.P.; Sakellaropoulos, N.; Ganz, T.; Nemeth, E. Hepcidin in iron overload disorders. Blood 2005, 105, 4103–4105. [Google Scholar]
- Malyszko, J. Hemojuvelin: The hepcidin story continues. Kidney Blood Press. Res 2009, 32, 71–76. [Google Scholar]
- Andriopoulos, B., Jr; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; Babitt, J.L. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar]
- Babitt, J.L.; Huang, F.W.; Xia, Y.; Sidis, Y.; Andrews, N.C.; Lin, H.Y. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J. Clin. Invest 2007, 117, 1933–1939. [Google Scholar]
- Casanovas, G.; Mleczko-Sanecka, K.; Altamura, S.; Hentze, M.W.; Muckenthaler, M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. (Berl.) 2009, 87, 471–480. [Google Scholar]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet 2009, 41, 478–481. [Google Scholar]
- Corradini, E.; Rozier, M.; Meynard, D.; Odhiambo, A.; Lin, H.Y.; Feng, Q.; Migas, M.C.; Britton, R.S.; Babitt, J.L.; Fleming, R.E. Iron regulation of hepcidin despite attenuated smad1,5,8 signaling in mice without transferrin receptor 2 or hfe. Gastroenterology 2011, 141, 1907–1914. [Google Scholar]
- Huang, F.W.; Pinkus, J.L.; Pinkus, G.S.; Fleming, M.D.; Andrews, N.C. A mouse model of juvenile hemochromatosis. J. Clin. Invest 2005, 115, 2187–2191. [Google Scholar]
- Niederkofler, V.; Salie, R.; Arber, S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Invest 2005, 115, 2180–2186. [Google Scholar]
- Wang, R.H.; Li, C.; Xu, X.; Zheng, Y.; Xiao, C.; Zerfas, P.; Cooperman, S.; Eckhaus, M.; Rouault, T.; Mishra, L.; Deng, C.X. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005, 2, 399–409. [Google Scholar]
- Camaschella, C.; Roetto, A.; Cali, A.; de Gobbi, M.; Garozzo, G.; Carella, M.; Majorano, N.; Totaro, A.; Gasparini, P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet 2000, 25, 14–15. [Google Scholar]
- Kawabata, H.; Yang, R.; Hirama, T.; Vuong, P.T.; Kawano, S.; Gombart, A.F.; Koeffler, H.P. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem 1999, 274, 20826–208232. [Google Scholar]
- Nemeth, E.; Roetto, A.; Garozzo, G.; Ganz, T.; Camaschella, C. Hepcidin is decreased in TFR2 hemochromatosis. Blood 2005, 105, 1803–1806. [Google Scholar]
- Wallace, D.F.; Summerville, L.; Lusby, P.E.; Subramaniam, V.N. First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut 2005, 54, 980–986. [Google Scholar]
- Chen, J.; Chloupkova, M.; Gao, J.; Chapman-Arvedson, T.L.; Enns, C.A. HFE modulates transferrin receptor 2 levels in hepatoma cells via interactions that differ from transferrin receptor 1-HFE interactions. J. Biol. Chem 2007, 282, 36862–36870. [Google Scholar]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab 2008, 7, 205–214. [Google Scholar]
- Waheed, A.; Britton, R.S.; Grubb, J.H.; Sly, W.S.; Fleming, R.E. HFE association with transferrin receptor 2 increases cellular uptake of transferrin-bound iron. Arch. Biochem. Biophys 2008, 474, 193–197. [Google Scholar]
- Fleming, R.E.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Koeffler, H.P.; Kawabata, H.; Britton, R.S.; Bacon, B.R.; Sly, W.S. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. USA 2002, 99, 10653–10658. [Google Scholar]
- Koyama, C.; Wakusawa, S.; Hayashi, H.; Ueno, T.; Suzuki, R.; Yano, M.; Saito, H.; Okazaki, T. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: Hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern. Med 2005, 44, 990–993. [Google Scholar]
- Majore, S.; Milano, F.; Binni, F.; Stuppia, L.; Cerrone, A.; Tafuri, A.; de Bernardo, C.; Palka, G.; Grammatico, P. Homozygous p.M172K mutation of the TFR2 gene in an Italian family with type 3 hereditary hemochromatosis and early onset iron overload. Haematologica 2006, 91, e92–e93. [Google Scholar]
- Mattman, A.; Huntsman, D.; Lockitch, G.; Langlois, S.; Buskard, N.; Ralston, D.; Butterfield, Y.; Rodrigues, P.; Jones, S.; Porto, G.; et al. Transferrin receptor 2 (TfR2) and HFE mutational analysis in non-C282Y iron overload: Identification of a novel TfR2 mutation. Blood 2002, 100, 1075–1077. [Google Scholar]
- Mayr, R.; Janecke, A.R.; Schranz, M.; Griffiths, W.J.; Vogel, W.; Pietrangelo, A.; Zoller, H. Ferroportin disease: A systematic meta-analysis of clinical and molecular findings. J. Hepatol 2010, 53, 941–949. [Google Scholar]
- Schimanski, L.M.; Drakesmith, H.; Merryweather-Clarke, A.T.; Viprakasit, V.; Edwards, J.P.; Sweetland, E.; Bastin, J.M.; Cowley, D.; Chinthammitr, Y.; Robson, K.J.; et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood 2005, 105, 4096–4102. [Google Scholar]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem 2000, 275, 19906–19912. [Google Scholar]
- Beutler, E. Hemochromatosis: Genetics and pathophysiology. Annu. Rev. Med 2006, 57, 331–347. [Google Scholar]
- Cremonesi, L.; Forni, G.L.; Soriani, N.; Lamagna, M.; Fermo, I.; Daraio, F.; Galli, A.; Pietra, D.; Malcovati, L.; Ferrari, M.; et al. Genetic and clinical heterogeneity of ferroportin disease. Br. J. Haematol 2005, 131, 663–670. [Google Scholar]
- Hetet, G.; Devaux, I.; Soufir, N.; Grandchamp, B.; Beaumont, C. Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations. Blood 2003, 102, 1904–1910. [Google Scholar]
- Kasvosve, I.; Gomo, Z.A.; Nathoo, K.J.; Matibe, P.; Mudenge, B.; Loyevsky, M.; Gordeuk, V.R. Effect of ferroportin Q248H polymorphism on iron status in African children. Am. J. Clin. Nutr 2005, 82, 1102–1106. [Google Scholar]
- Aguilar-Martinez, P.; Schved, J.F.; Brissot, P. The evaluation of hyperferritinemia: An updated strategy based on advances in detecting genetic abnormalities. Am. J. Gastroenterol 2005, 100, 1185–1194. [Google Scholar]
- Brissot, P.; Troadec, M.B.; Bardou-Jacquet, E.; Le Lan, C.; Jouanolle, A.M.; Deugnier, Y.; Loreal, O. Current approach to hemochromatosis. Blood Rev 2008, 22, 195–210. [Google Scholar]
- Le Gac, G.; Dupradeau, F.Y.; Mura, C.; Jacolot, S.; Scotet, V.; Esnault, G.; Mercier, A.Y.; Rochette, J.; Ferec, C. Phenotypic expression of the C282Y/Q283P compound heterozygosity in HFE and molecular modeling of the Q283P mutation effect. Blood Cells Mol. Dis 2003, 30, 231–237. [Google Scholar]
- Brissot, P.; Troadec, M.B.; Loreal, O. The clinical relevance of new insights in iron transport and metabolism. Curr. Hematol. Rep 2004, 3, 107–115. [Google Scholar]
- EASL clinical practice guidelines for HFE hemochromatosis. J. Hepatol. 2010, 53, 3–22.
- Brissot, P.; de Bels, F. Current approaches to the management of hemochromatosis. Hematol. Am. Soc. Hematol. Educ. Program 2006, 36–41. [Google Scholar]
- Van Bokhoven, M.A.; van Deursen, C.T.; Swinkels, D.W. Diagnosis and management of hereditary haemochromatosis. BMJ 2011, 342. [Google Scholar] [CrossRef]
- de Juan Jimenez, I.; Cardenosa, E.E.; Suela, S.P.; Gonzalez, E.B.; Trejo, D.S.; Lluch, O.F.; Gilabert, P.B. Advantage of high-resolution melting curve analysis over conformation-sensitive gel electrophoresis for mutational screening of BRCA1 and BRCA2 genes. Clin. Chim. Acta 2011, 412, 578–582. [Google Scholar]
- Santos, P.C.; Soares, R.A.; Krieger, J.E.; Guerra-Shinohara, E.M.; Pereira, A.C. Genotyping of the hemochromatosis HFE p.H63D and p.C282Y mutations by high-resolution melting with the Rotor-Gene 6000((R)) instrument. Clin. Chem. Lab. Med 2011, 49, 1633–1636. [Google Scholar]
- Tag, C.G.; Gressner, A.M.; Weiskirchen, R. An unusual melting curve profile in LightCycler multiplex genotyping of the hemochromatosis H63D/C282Y gene mutations. Clin. Biochem 2001, 34, 511–515. [Google Scholar]
- Castiglioni, E.; Soriani, N.; Girelli, D.; Camaschella, C.; Spiga, I.; Della Porta, M.G.; Ferrari, M.; Cremonesi, L. High resolution melting for the identification of mutations in the iron responsive element of the ferritin light chain gene. Clin. Chem. Lab. Med 2011, 48, 1415–1418. [Google Scholar]
- Lin, J.T.; Hsiao, K.J.; Chen, C.Y.; Wu, C.C.; Lin, S.J.; Chou, Y.Y.; Shiesh, S.C. High resolution melting analysis for the detection of SLC25A13 gene mutations in Taiwan. Clin. Chim. Acta 2011, 412, 460–465. [Google Scholar]



{kind=link}
{kind=link}
| HH types | Phenotype MIM number | Gene MIM number | Location | Inheritance | Gene product function | Main clinical manifestations |
|---|---|---|---|---|---|---|
| 1 | 235200 | HFE, 613609 | 6p21.3 | AR | Involved in hepcidin synthesis via BMP6, interaction with TFR1. | Arthropathy, skin pigmentation, liver damage, diabetes, endocrine dysfunction, cardiomyopathy, hypogonadism. |
| 2A | 602390 | HJV, 608374 | 1p21 | AR | Involved in hepcidin synthesis, BMP co-receptor. | Types 2: earlier onset, <30 years old. Hypogonadism and cardiomyopathy more prevalent. |
| 2B | 613313 | HAMP, 606464 | 19q13 | AR | Downregulation of iron efflux from enterocytes. | |
| 3 | 604250 | TFR2, 604720 | 7q22 | AR | Involved in hepcidin synthesis, interaction with transferrin. | As for type 1. |
| 4 | 606069 | SLC40A1, 604653 | 2q32 | AD | Duodenal iron export. | Lower tolerance to phlebotomies and may have anemia. |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Santos, P.C.J.L.; Krieger, J.E.; Pereira, A.C. Molecular Diagnostic and Pathogenesis of Hereditary Hemochromatosis. Int. J. Mol. Sci. 2012, 13, 1497-1511. https://doi.org/10.3390/ijms13021497
Santos PCJL, Krieger JE, Pereira AC. Molecular Diagnostic and Pathogenesis of Hereditary Hemochromatosis. International Journal of Molecular Sciences. 2012; 13(2):1497-1511. https://doi.org/10.3390/ijms13021497
Chicago/Turabian StyleSantos, Paulo C. J. L., Jose E. Krieger, and Alexandre C. Pereira. 2012. "Molecular Diagnostic and Pathogenesis of Hereditary Hemochromatosis" International Journal of Molecular Sciences 13, no. 2: 1497-1511. https://doi.org/10.3390/ijms13021497
APA StyleSantos, P. C. J. L., Krieger, J. E., & Pereira, A. C. (2012). Molecular Diagnostic and Pathogenesis of Hereditary Hemochromatosis. International Journal of Molecular Sciences, 13(2), 1497-1511. https://doi.org/10.3390/ijms13021497