Using Support Vector Machine and Evolutionary Profiles to Predict Antifreeze Protein Sequences

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Protein Features and Vector Encoding
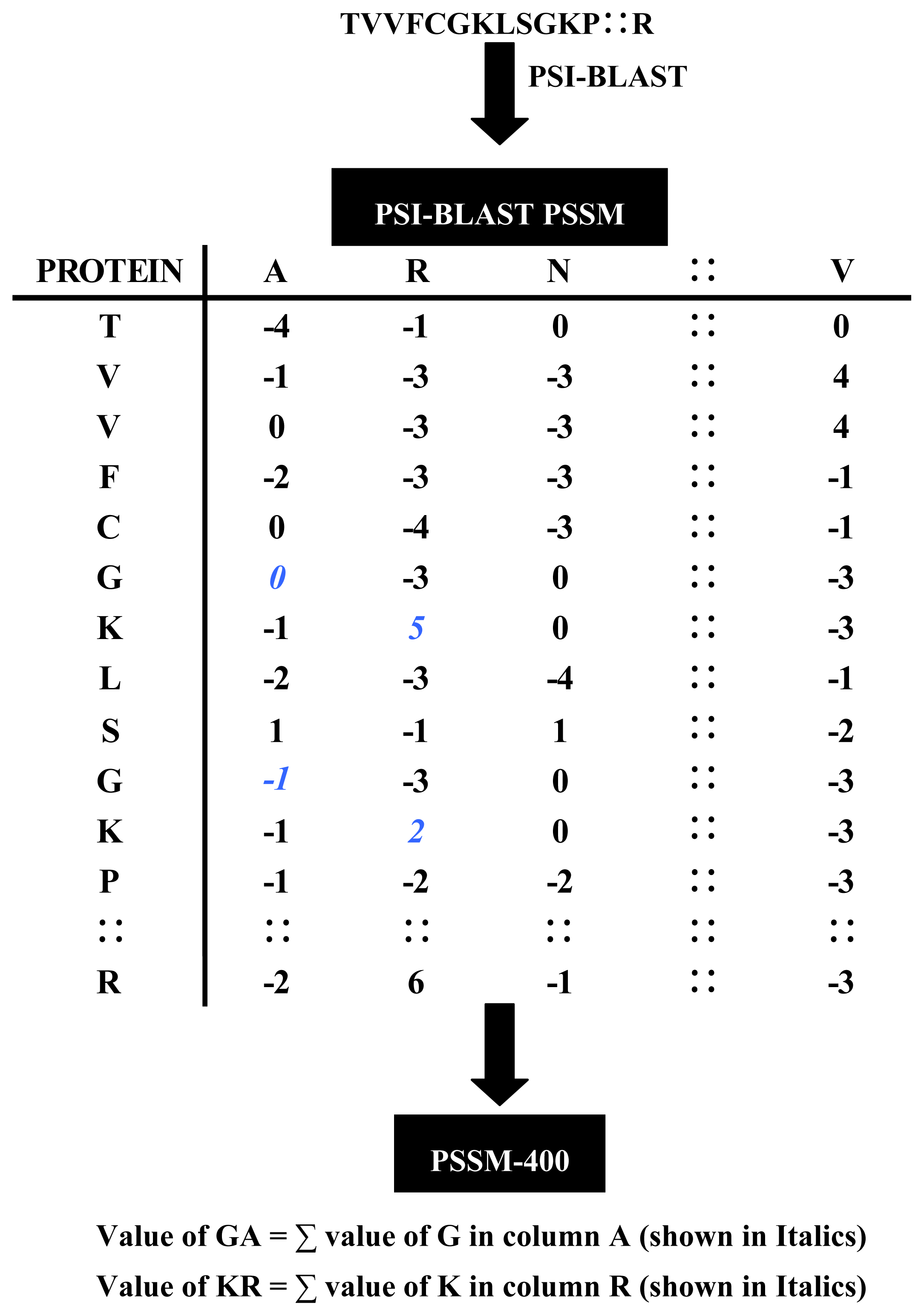
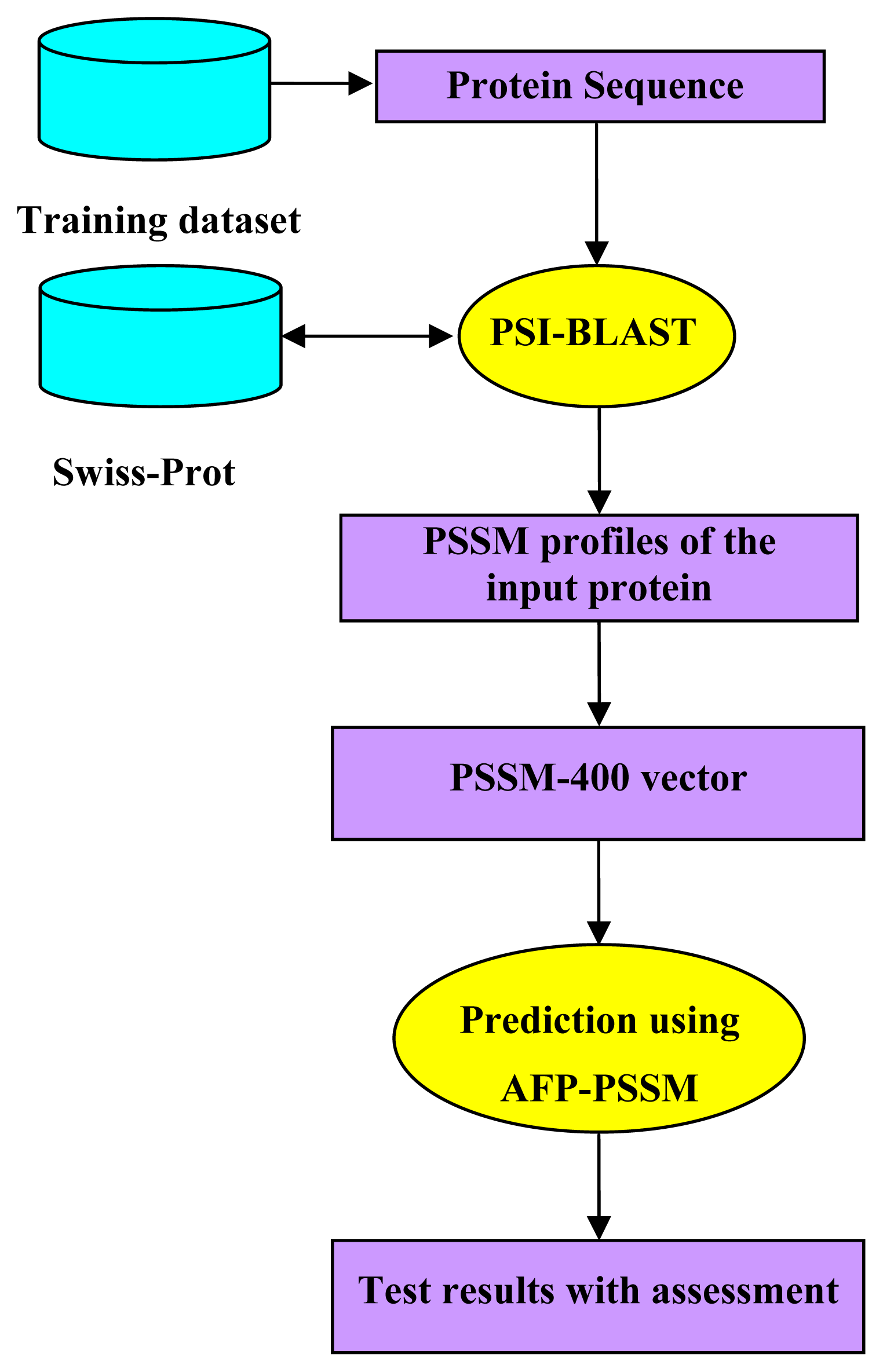
2.2.1. Evolutionary Information
2.2.2. Amino Acid and Dipeptide Composition
2.2.3. Chou’s Pseudo Amino acid Composition
2.3. Support Vector Machines
2.4. Evaluation
2.5. Model Building and Protocol Guide
3. Results and Discussion
4. Conclusions
Supplementary Information
ijms-13-02196-s001.pdfAcknowledgments
References
- Davies, P.L.; Baardsnes, J.; Kuiper, M.J.; Walker, V.K. Structure and function of antifreeze proteins. Philos. Trans. R. Soc. Lond. B 2002, 357, 927–935. [Google Scholar]
- Sformo, T.; Kohl, F.; McIntyre, J.; Kerr, P.; Duman, J.G.; Barnes, B.M. Simultaneous freeze tolerance and avoidance in individual fungus gnats, Exechia nugatoria. J. Comp. Physiol. B 2009, 179, 897–902. [Google Scholar]
- Lewitt, J. Responses of Plants to Environmental Stresses; Academic Press: New York NY, USA, 1980; Volume 1, pp. 269–295. [Google Scholar]
- Scholander, P.F.; van Dam, L.; Kanwisher, J.W.; Hammel, H.T.; Gordon, M.S. Supercooling and osmoregulation in arctic fish. J. Cell. Comp. Physiol 1957, 49, 5–24. [Google Scholar]
- Moriyama, M.; Abe, J.; Yoshida, M.; Tsurumi, Y.; Nakayama, S. Seasonal changes in freezing tolerance, moisture content and dry weight of three temperate grasses. Grassl. Sci 1995, 41, 21–25. [Google Scholar]
- Logsdon, J.M.; Doolittle, W.F. Origin of antifreeze protein genes: A cool tale in molecular evolution. Proc. Natl. Acad. Sci. USA 1997, 94, 3485–3487. [Google Scholar]
- Ewart, K.V.; Lin, Q.; Hew, C.L. Structure, function and evolution of antifreeze proteins. Cell. Mol. Life Sci 1999, 55, 271–283. [Google Scholar]
- Davies, P.L.; Baardsnes, J.; Kuiper, M.J.; Walker, V.K. Structure and function of antifreeze proteins. Philos. Trans. R. Soc. Lond. B 2002, 357, 927–935. [Google Scholar]
- Davies, P.L.; Sykes, B.D. Antifreeze proteins. Curr. Opin. Struct. Biol 1997, 7, 828–834. [Google Scholar]
- Cheng, C.H. Evolution of the diverse antifreeze proteins. Curr. Opin. Genet. Dev 1998, 8, 715–720. [Google Scholar]
- Urrutia, M.E.; Duman, J.G.; Knight, C.A. Plant thermal hysteresis proteins. Biochim. Biophys. Acta 1992, 1121, 199–206. [Google Scholar]
- Yu, X.M.; Griffith, M. Winter rye antifreeze activity increases in response to cold and drought, but not abscisic acid. Physiol. Plant 2001, 112, 78–86. [Google Scholar]
- Griffith, M.; Ewart, K.V. Antifreeze proteins and their potential use in frozen foods. Biotechnol. Adv 1995, 13, 375–402. [Google Scholar]
- Breton, G.; Danyluk, J.; Ouellet, F.; Sarhan, F. Biotechnological applications of plant freezing associated proteins. Biotechnol. Annu. Rev 2000, 6, 59–101. [Google Scholar]
- Shao, X.; Tian, Y.; Wu, L.; Wang, Y.; Jing, L.; Deng, N. Predicting DNA- and RNA-binding proteins from sequences with kernel methods. J. Theor. Biol 2009, 258, 289–293. [Google Scholar]
- Chou, K.C.; Shen, H.B. Review: Recent progresses in protein subcellular location prediction. Anal. Biochem 2007, 370, 1–16. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc 2008, 3, 153–162. [Google Scholar]
- Chou, K.C. Review: Structural bioinformatics and its impact to biomedical science. Curr. Med. Chem 2004, 11, 2105–2134. [Google Scholar]
- Chou, K.C.; Shen, H.B. ProtIdent: A web server for identifying proteases and their types by fusing functional domain and sequential evolution information. Biochem. Biophys. Res. Commun 2008, 376, 321–325. [Google Scholar]
- Chou, K.C.; Shen, H.B. MemType-2L: A web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem. Biophys. Res. Commun 2007, 360, 339–345. [Google Scholar]
- Chou, K.C.; Wei, D.Q.; Zhong, W.Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun 2003, 308, 148–151. [Google Scholar]
- Li, Y.; Wei, D.Q.; Gao, W.N.; Gao, H.; Liu, B.N.; Huang, C.J.; Xu, W.R.; Liu, D.K.; Chen, H.F.; Chou, K.C. Computational approach to drug design for oxazolidinones as antibacterial agents. Med. Chem 2007, 3, 576–582. [Google Scholar]
- Wang, J.F.; Wei, D.Q.; Chen, C.; Li, Y.; Chou, K.C. Molecular modeling of two CYP2C19 SNPs and its implications for personalized drug design. Protein Pept. Lett 2008, 15, 27–32. [Google Scholar]
- Shen, H.B.; Chou, K.C. EzyPred: A top-down approach for predicting enzyme functional classes and subclasses. Biochem. Biophys. Res. Commun 2007, 364, 53–59. [Google Scholar]
- Chou, K.C.; Shen, H.B. Signal-CF: A subsite-coupled and window-fusing approach for predicting signal peptides. Biochem. Biophys. Res. Commun 2007, 357, 633–640. [Google Scholar]
- Shen, H.B.; Chou, K.C. Signal-3L: A 3-layer approach for predicting signal peptide. Biochem. Biophys. Res. Commun 2007, 363, 297–303. [Google Scholar]
- Kandaswamy, K.K.; Chou, K.; Martinetz, T.; Möller, S.; Suganthan, P.N.; Sridharan, S.; Ganesan, P. AFP-Pred: A random forest approach for predicting antifreeze proteins from sequence-derived properties. J. Theor. Biol 2011, 270, 56–62. [Google Scholar]
- AFP_PSSM. Available online: http://59.73.198.144/AFP_PSSM/ accessed on 28 December 2011.
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J. Theor. Biol 2011, 273, 236–247. [Google Scholar]
- Li, W.; Jaroszewski, L.; Odzik, G.A. Clustering of highly homologous sequences to reduce the size of large protein database. Bioinformatics 2001, 17, 282–283. [Google Scholar]
- AFP-dataset. Available online: http://www3.ntu.edu.sg/home/EPNSugan/index_files/AFP_Pred.htm accessed on 16 June 2011.
- Zhao, X.W.; Li, X.T.; Ma, Z.Q.; Yin, M.H. Prediction of lysine ubiquitylation with ensemble classifier and feature selection. Int. J. Mol. Sci 2011, 12, 8347–8361. [Google Scholar]
- Jones, D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 2007, 23, 538–544. [Google Scholar]
- Ruchi, V.; Grish, C.V.; Raghava, G.P.S. Prediction of mitochondrial proteins of malaria parasite using split amino acid composition and PSSM profile. Amino Acids 2010, 39, 101–110. [Google Scholar]
- Schaffer, A.; Aravind, L.; Madden, T.; Shavirin, S.; Spouge, J.; Wolf, Y.; Koonin, E.; Altschul, S. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 2001, 29, 2994–3005. [Google Scholar]
- Altschul, S.; Madden, T.; Schaffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Swiss-Prot database. Available online: http://www.uniprot.org/uniprot/?query=reviewed%3Ayes accessed on 16 October 2011.
- Kumar, M.; Gromiha, M.M.; Raghava, G.P.S. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinforma 2007, 8. [Google Scholar] [CrossRef]
- Mohabatkar, H. Prediction of cyclin proteins using Chou’s pseudo amino acid composition. Protein Pept. Lett 2010, 17, 1207–1214. [Google Scholar]
- Esmaeili, M.; Mohabatkar, H.; Mohsenzadeh, S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J. Theor. Biol 2010, 263, 203–209. [Google Scholar]
- Lin, H. The modified Mahalanobis discriminant for predicting outer membrane proteins by using Chou’s pseudo amino acid composition. J. Theor. Biol 2008, 252, 350–356. [Google Scholar]
- Zeng, Y.H.; Guo, Y.Z.; Xiao, R.Q.; Yang, L.; Yu, L.Z.; Li, M.L. Using the augmented Chou’s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach. J. Theor. Biol 2009, 259, 366–372. [Google Scholar]
- Chen, C.; Chen, L.; Zou, X.; Cai, P. Prediction of protein secondary structure content by using the concept of Chou’s pseudo amino acid composition and support vector machine. Protein Pept. Lett 2009, 16, 27–31. [Google Scholar]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res 2008, 36, D202–D205. [Google Scholar]
- Vapnik, V. Statistical Learning Theory; Wiley-Interscience: New York, NY, USA, 1998. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machine 2001, 2. [CrossRef]
- LIBSVM. Available online: http://www.csite.ntu.edu.tw/~sjlin/libsvm accessed on 23 June 2011.
- Chou, K.C.; Zhang, C.T. Review: Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol 1995, 30, 275–349. [Google Scholar]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc 2008, 3, 153–162. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
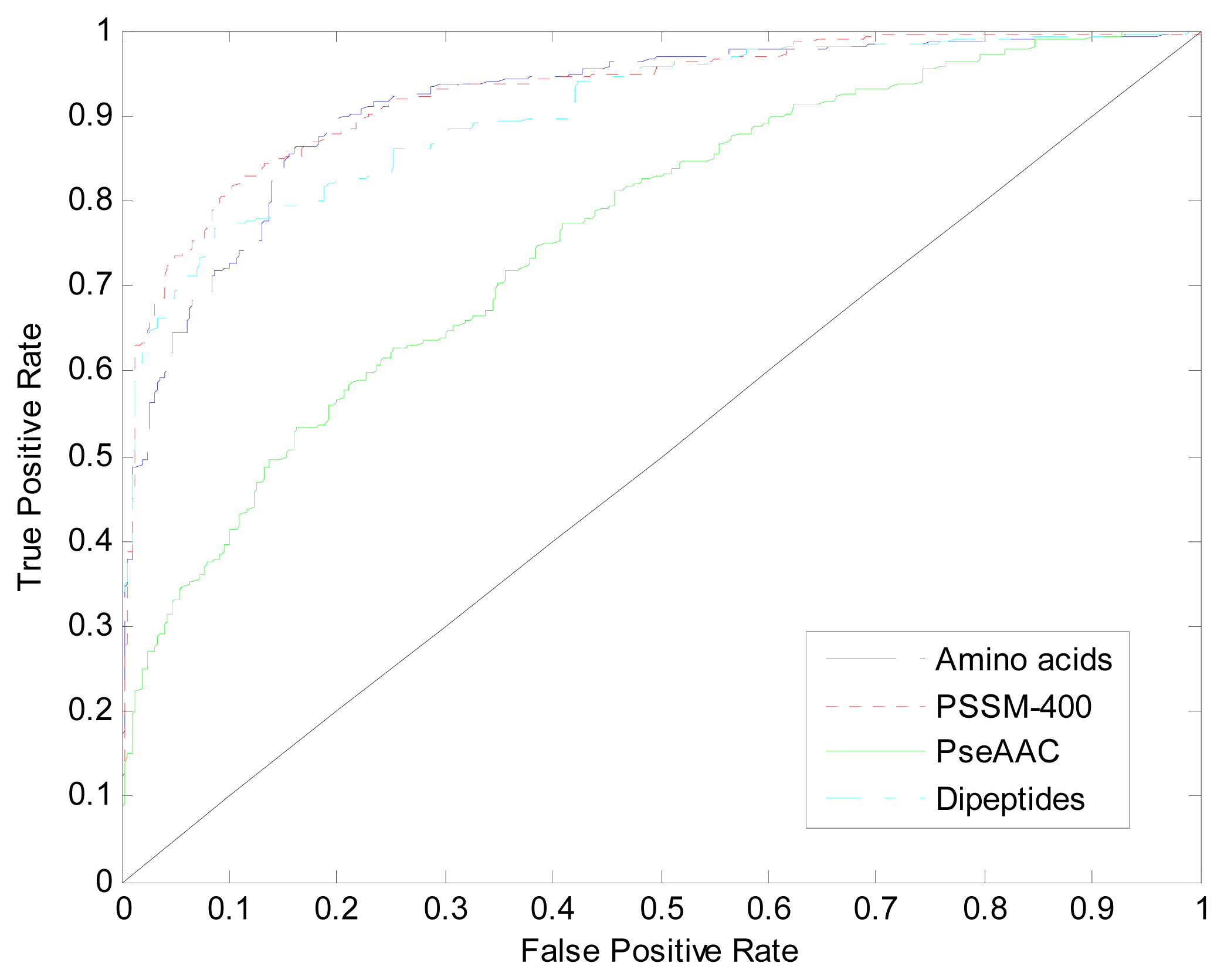
| Method | Amino Acids | Dipeptides | PseAAC | PSSM-400 |
|---|---|---|---|---|
| Acc | 80.83% | 78.83% | 56.18% | 82.67% |
| AUC | 0.912 | 0.904 | 0.761 | 0.926 |
| Method | Sn (%) | Sp (%) | Acc (%) |
|---|---|---|---|
| AFP-Pred [27] | 84.67 | 82.32 | 83.38 |
| AFP_PSSM | 75.89 | 93.28 | 93.01 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhao, X.; Ma, Z.; Yin, M. Using Support Vector Machine and Evolutionary Profiles to Predict Antifreeze Protein Sequences. Int. J. Mol. Sci. 2012, 13, 2196-2207. https://doi.org/10.3390/ijms13022196
Zhao X, Ma Z, Yin M. Using Support Vector Machine and Evolutionary Profiles to Predict Antifreeze Protein Sequences. International Journal of Molecular Sciences. 2012; 13(2):2196-2207. https://doi.org/10.3390/ijms13022196
Chicago/Turabian StyleZhao, Xiaowei, Zhiqiang Ma, and Minghao Yin. 2012. "Using Support Vector Machine and Evolutionary Profiles to Predict Antifreeze Protein Sequences" International Journal of Molecular Sciences 13, no. 2: 2196-2207. https://doi.org/10.3390/ijms13022196
APA StyleZhao, X., Ma, Z., & Yin, M. (2012). Using Support Vector Machine and Evolutionary Profiles to Predict Antifreeze Protein Sequences. International Journal of Molecular Sciences, 13(2), 2196-2207. https://doi.org/10.3390/ijms13022196