Phage Displayed Peptides/Antibodies Recognizing Growth Factors and Their Tyrosine Kinase Receptors as Tools for Anti-Cancer Therapeutics

 ,
, 
Abstract
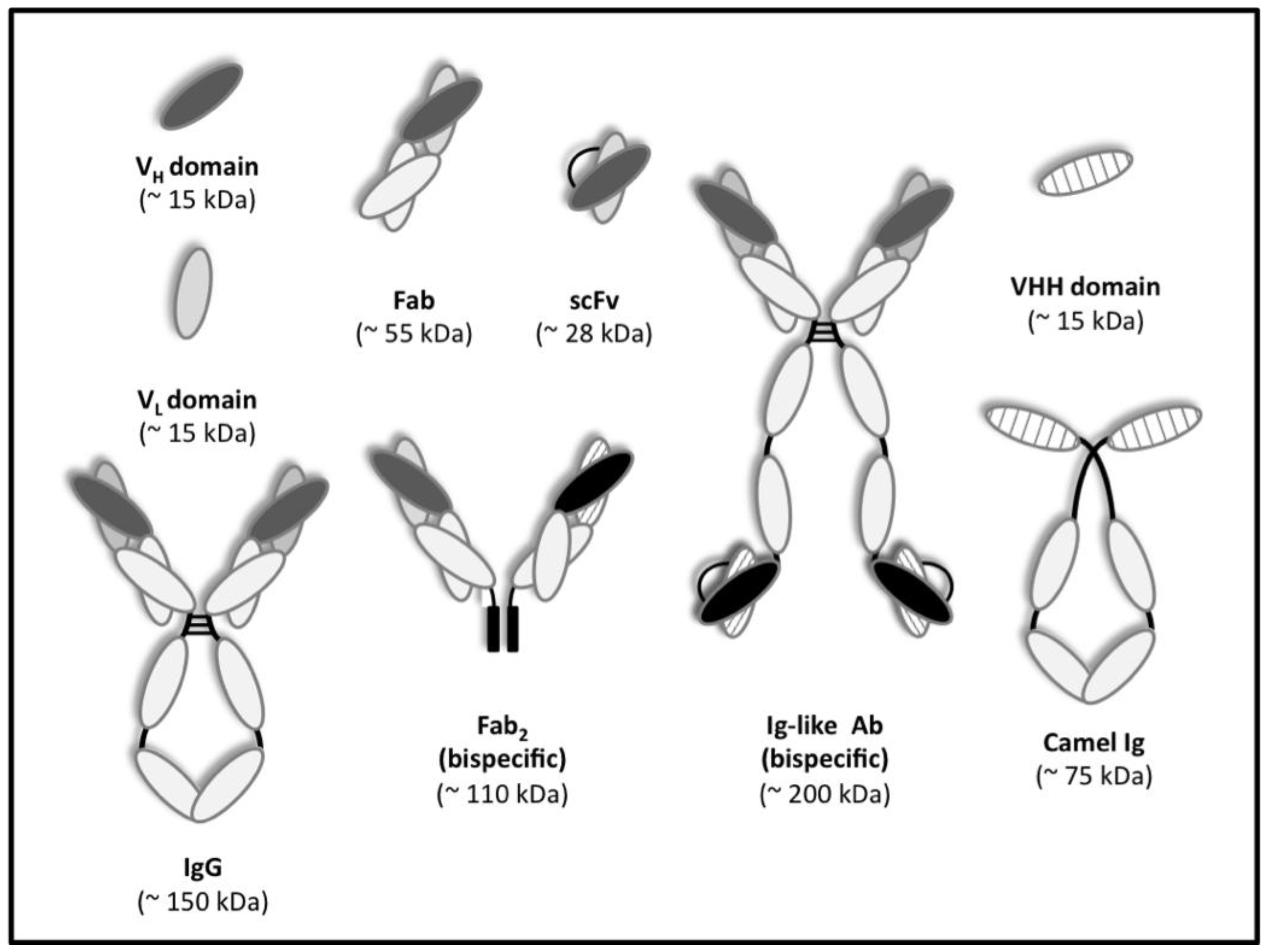
:1. Antibody Display
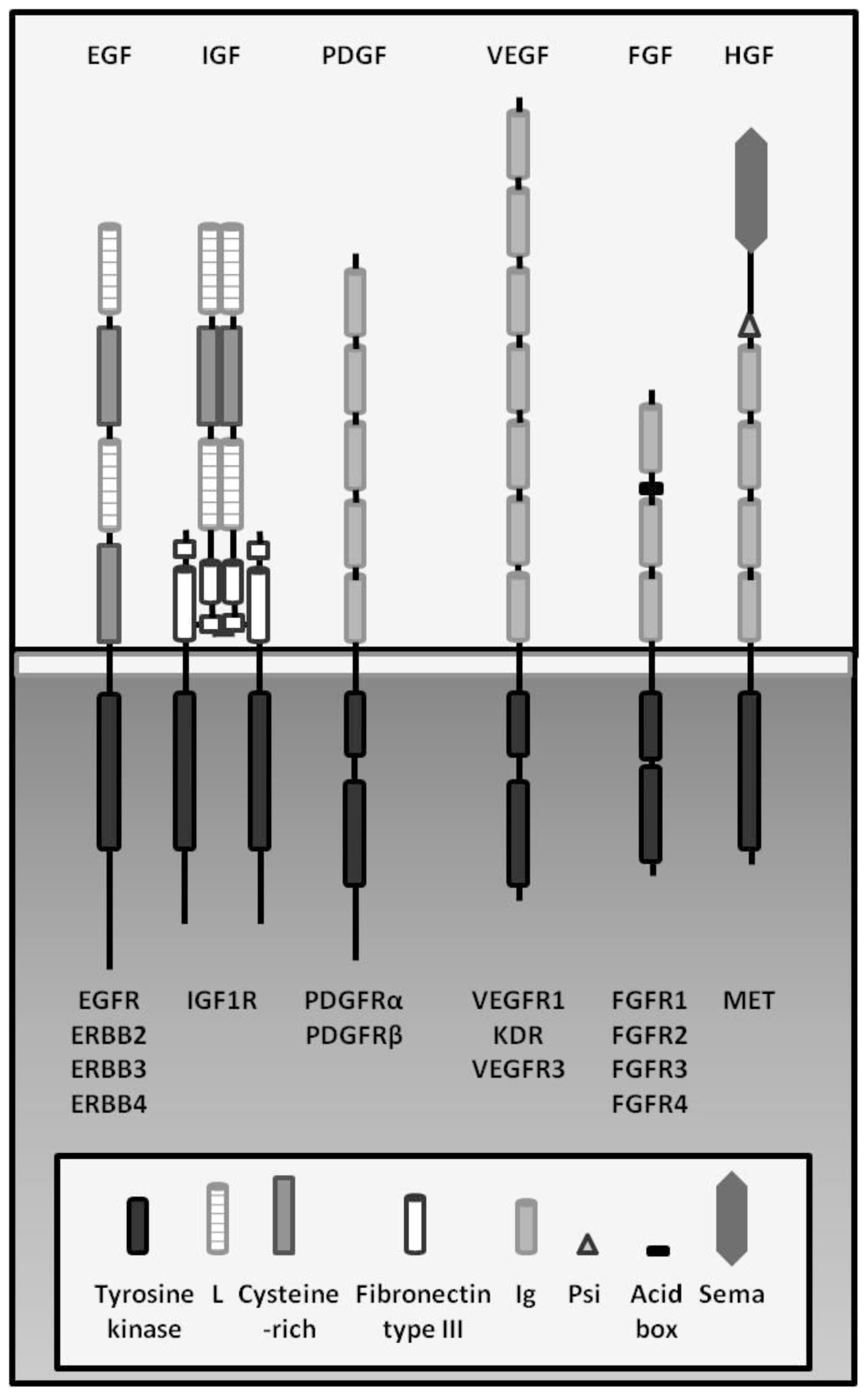
2. Growth Factors and Cancer
2.1. EGF Family/ERBB Family
2.2. HGF/MET
2.3. IGFs/IGFRs
2.4. VEGFs/VEGFRs
2.5. FGFs/FGFRs
3. Preclinical Studies
3.1. Phages Displaying Peptides
3.2. Phages Displaying Antibodies
4. Clinical Studies
4.1. IMC-A12 (Cixutumumab)
4.2. IMC-11F8 (Necitumumab)
4.3. IMC-1121b (Ramucirumab)
4.4. IMC-3C5
4.5. AMG-479 (Ganitumab)
4.6. VGX-100 (Fresolimumab)
Acknowledgments
References
- Edelman, G.M. Antibody structure and molecular immunology. Science 1973, 180, 830–840. [Google Scholar]
- Davies, D.R.; Cohen, G.H. Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. USA 1996, 93, 7–12. [Google Scholar]
- Skerra, A.; Pluckthun, A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science 1988, 240, 1038–1041. [Google Scholar]
- Orlandi, R.; Gussow, D.H.; Jones, P.T.; Winter, G. Cloning immunoglobulin variable domains for expression by the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 3833–3837. [Google Scholar]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar]
- Barbas, C.F., 3rd; Kang, A.S.; Lerner, R.A.; Benkovic, S.J. Assembly of combinatorial antibody libraries on phage surfaces: The gene III site. Proc. Natl. Acad. Sci. USA 1991, 88, 7978–7982. [Google Scholar]
- Watkins, N.A.; Ouwehand, W.H. Introduction to antibody engineering and phage display. Vox Sang 2000, 78, 72–79. [Google Scholar]
- Engberg, J.; Andersen, P.S.; Nielsen, L.K.; Dziegiel, M.; Johansen, L.K.; Albrechtsen, B. Phage-display libraries of murine and human antibody Fab fragments. Mol. Biotechnol 1996, 6, 287–310. [Google Scholar]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol 1991, 222, 581–597. [Google Scholar]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev 1997, 97, 391–410. [Google Scholar]
- Georgieva, Y.; Konthur, Z. Design and screening of M13 phage display cDNA libraries. Molecules 2011, 16, 1667–1681. [Google Scholar]
- Iannolo, G.; Minenkova, O.; Petruzzelli, R.; Cesareni, G. Modifying filamentous phage capsid: Limits in the size of the major capsid protein. J. Mol. Biol 1995, 248, 835–844. [Google Scholar]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv 2010, 28, 849–858. [Google Scholar]
- Sidhu, S.S. Engineering M13 for phage display. Biomol. Eng 2001, 18, 57–63. [Google Scholar]
- Azzazy, H.M.; Highsmith, W.E., Jr. Phage display technology: Clinical applications and recent innovations. Clin. Biochem 2002, 35, 425–445. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol 2005, 23, 1126–1136. [Google Scholar]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Cheng, N.; Chytil, A.; Shyr, Y.; Joly, A.; Moses, H.L. Transforming growth factor-beta signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol. Cancer Res 2008, 6, 1521–1533. [Google Scholar]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar]
- Forbes, S.A.; Tang, G.; Bindal, N.; Bamford, S.; Dawson, E.; Cole, C.; Kok, C.Y.; Jia, M.; Ewing, R.; Menzies, A.; et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res 2010, 38, D652–D657. [Google Scholar]
- Takahashi, J.A.; Fukumoto, M.; Igarashi, K.; Oda, Y.; Kikuchi, H.; Hatanaka, M. Correlation of basic fibroblast growth factor expression levels with the degree of malignancy and vascularity in human gliomas. J. Neurosurg 1992, 76, 792–798. [Google Scholar]
- Humphrey, P.A.; Gangarosa, L.M.; Wong, A.J.; Archer, G.E.; Lund-Johansen, M.; Bjerkvig, R.; Laerum, O.D.; Friedman, H.S.; Bigner, D.D. Deletion-mutant epidermal growth factor receptor in human gliomas: Effects of type II mutation on receptor function. Biochem. Biophys. Res. Commun 1991, 178, 1413–1420. [Google Scholar]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar]
- Jenny, B.; Harrison, J.A.; Baetens, D.; Tille, J.C.; Burkhardt, K.; Mottaz, H.; Kiss, J.Z.; Dietrich, P.Y.; de Tribolet, N.; Pizzolato, G.P.; et al. Expression and localization of VEGF-C and VEGFR-3 in glioblastomas and haemangioblastomas. J. Pathol 2006, 209, 34–43. [Google Scholar]
- Baselga, J.; Arteaga, C.L. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J. Clin. Oncol 2005, 23, 2445–2459. [Google Scholar]
- Seiwert, T.Y.; Jagadeeswaran, R.; Faoro, L.; Janamanchi, V.; Nallasura, V.; El Dinali, M.; Yala, S.; Kanteti, R.; Cohen, E.E.; Lingen, M.W.; et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res 2009, 69, 3021–3031. [Google Scholar]
- Marshall, M.E.; Hinz, T.K.; Kono, S.A.; Singleton, K.R.; Bichon, B.; Ware, K.E.; Marek, L.; Frederick, B.A.; Raben, D.; Heasley, L.E. Fibroblast growth factor receptors are components of autocrine signaling networks in head and neck squamous cell carcinoma cells. Clin. Cancer Res 2011, 17, 5016–5025. [Google Scholar]
- Lin, W.; Kao, H.W.; Robinson, D.; Kung, H.J.; Wu, C.W.; Chen, H.C. Tyrosine kinases and gastric cancer. Oncogene 2000, 19, 5680–5689. [Google Scholar]
- Zhao, Z.S.; Zhou, J.L.; Yao, G.Y.; Ru, G.Q.; Ma, J.; Ruan, J. Correlative studies on bFGF mRNA and MMP-9 mRNA expressions with microvascular density, progression, and prognosis of gastric carcinomas. World J. Gastroenterol 2005, 11, 3227–3233. [Google Scholar]
- Barclay, C.; Li, A.W.; Geldenhuys, L.; Baguma-Nibasheka, M.; Porter, G.A.; Veugelers, P.J.; Murphy, P.R.; Casson, A.G. Basic fibroblast growth factor (FGF-2) overexpression is a risk factor for esophageal cancer recurrence and reduced survival, which is ameliorated by coexpression of the FGF-2 antisense gene. Clin Cancer Res 2005, 11, 7683–7691. [Google Scholar]
- Spratlin, J.L.; Cohen, R.B.; Eadens, M.; Gore, L.; Camidge, D.R.; Diab, S.; Leong, S.; O’Bryant, C.; Chow, L.Q.; Serkova, N.J.; et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J. Clin. Oncol 2010, 28, 780–787. [Google Scholar]
- Gao, J.; Inagaki, Y.; Song, P.; Qu, X.; Kokudo, N.; Tang, W. Targeting c-Met as a promising strategy for the treatment of hepatocellular carcinoma. Pharmacol. Res 2012, 65, 23–30. [Google Scholar]
- Golan, T.; Javle, M. Targeting the insulin growth factor pathway in gastrointestinal cancers. Oncology (Williston Park) 2011, 25. [Google Scholar]
- Reidy, D.L.; Vakiani, E.; Fakih, M.G.; Saif, M.W.; Hecht, J.R.; Goodman-Davis, N.; Hollywood, E.; Shia, J.; Schwartz, J.; Chandrawansa, K.I. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J. Clin. Oncol 2010, 28, 4240–4246. [Google Scholar]
- Essapen, S.; Thomas, H.; Green, M.; de Vries, C.; Cook, M.G.; Marks, C.; Topham, C.; Modjtahedi, H. The expression and prognostic significance of HER-2 in colorectal cancer and its relationship with clinicopathological parameters. Int. J. Oncol 2004, 24, 241–248. [Google Scholar]
- Kammula, U.S.; Kuntz, E.J.; Francone, T.D.; Zeng, Z.; Shia, J.; Landmann, R.G.; Paty, P.B.; Weiser, M.R. Molecular co-expression of the c-Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett 2007, 248, 219–228. [Google Scholar]
- Kos, M.; Dabrowski, A. Tumour’s angiogenesis—The function of VEGF and bFGF in colorectal cancer. Ann. Univ. Mariae Curie Sklodowska Med 2002, 57, 556–561. [Google Scholar]
- Volm, M.; Koomagi, R.; Mattern, J.; Stammler, G. Prognostic value of basic fibroblast growth factor and its receptor (FGFR-1) in patients with non-small cell lung carcinomas. Eur. J. Cancer 1997, 33, 691–693. [Google Scholar]
- Katoh, M. Cancer genomics and genetics of FGFR2 (review). Int. J. Oncol 2008, 33, 233–237. [Google Scholar]
- Rusch, V.; Baselga, J.; Cordon-Cardo, C.; Orazem, J.; Zaman, M.; Hoda, S.; McIntosh, J.; Kurie, J.; Dmitrovsky, E. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res 1993, 53, 2379–2385. [Google Scholar]
- Horstmann, M.; Merseburger, A.S.; von der Heyde, E.; Serth, J.; Wegener, G.; Mengel, M.; Feil, G.; Hennenlotter, J.; Nagele, U.; Anastasiadis, A.; et al. Correlation of bFGF expression in renal cell cancer with clinical and histopathological features by tissue microarray analysis and measurement of serum levels. J. Cancer Res. Clin. Oncol 2005, 131, 715–722. [Google Scholar]
- Banumathy, G.; Cairns, P. Signaling pathways in renal cell carcinoma. Cancer Biol. Ther 2010, 10, 658–664. [Google Scholar]
- Yuen, J.S.; Cockman, M.E.; Sullivan, M.; Protheroe, A.; Turner, G.D.; Roberts, I.S.; Pugh, C.W.; Werner, H.; Macaulay, V.M. The VHL tumor suppressor inhibits expression of the IGF1R and its loss induces IGF1R upregulation in human clear cell renal carcinoma. Oncogene 2007, 26, 6499–6508. [Google Scholar]
- Kojima, S.; Inahara, M.; Suzuki, H.; Ichikawa, T.; Furuya, Y. Implications of insulin-like growth factor-I for prostate cancer therapies. Int. J. Urol 2009, 16, 161–167. [Google Scholar]
- Neto, A.S.; Tobias-Machado, M.; Wroclawski, M.L.; Fonseca, F.L.; Pompeo, A.C.; del Giglio, A. Molecular oncogenesis of prostate adenocarcinoma: Role of the human epidermal growth factor receptor 2 (HER-2/neu). Tumori 2010, 96, 645–649. [Google Scholar]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 2004, 11, 709–724. [Google Scholar]
- Gravdal, K.; Halvorsen, O.J.; Haukaas, S.A.; Akslen, L.A. Expression of bFGF/FGFR-1 and vascular proliferation related to clinicopathologic features and tumor progress in localized prostate cancer. Virchows Arch 2006, 448, 68–74. [Google Scholar]
- Knowles, M.A. Novel therapeutic targets in bladder cancer: Mutation and expression of FGF receptors. Future Oncol 2008, 4, 71–83. [Google Scholar]
- Black, P.C.; Dinney, C.P. Growth factors and receptors as prognostic markers in urothelial carcinoma. Curr. Urol. Rep 2008, 9, 55–61. [Google Scholar]
- Van Dam, P.A.; Vergote, I.B.; Lowe, D.G.; Watson, J.V.; van Damme, P.; van der Auwera, J.C.; Shepherd, J.H. Expression of c-erbB-2, c-myc, and c-ras oncoproteins, insulin-like growth factor receptor I, and epidermal growth factor receptor in ovarian carcinoma. J. Clin. Pathol 1994, 47, 914–919. [Google Scholar]
- Wang, S.C.; Hung, M.C. HER2 overexpression and cancer targeting. Semin. Oncol 2001, 28, 115–124. [Google Scholar]
- Jin, Q.; Esteva, F.J. Cross-talk between the ErbB/HER family and the type I insulin-like growth factor receptor signaling pathway in breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 485–498. [Google Scholar]
- Penault-Llorca, F.; Bertucci, F.; Adelaide, J.; Parc, P.; Coulier, F.; Jacquemier, J.; Birnbaum, D.; deLapeyriere, O. Expression of FGF and FGF receptor genes in human breast cancer. Int. J. Cancer 1995, 61, 170–176. [Google Scholar]
- Marsh, S.K.; Bansal, G.S.; Zammit, C.; Barnard, R.; Coope, R.; Roberts-Clarke, D.; Gomm, J.J.; Coombes, R.C.; Johnston, C.L. Increased expression of fibroblast growth factor 8 in human breast cancer. Oncogene 1999, 18, 1053–1060. [Google Scholar]
- Easty, D.J.; Gray, S.G.; O’Byrne, K.J.; O’Donnell, D.; Bennett, D.C. Receptor tyrosine kinases and their activation in melanoma. Pigm. Cell Melanoma Res 2011, 24, 446–461. [Google Scholar]
- Kornmann, M.; Beger, H.G.; Korc, M. Role of fibroblast growth factors and their receptors in pancreatic cancer and chronic pancreatitis. Pancreas 1998, 17, 169–175. [Google Scholar]
- Scotlandi, K.; Picci, P. Targeting insulin-like growth factor 1 receptor in sarcomas. Curr. Opin. Oncol 2008, 20, 419–427. [Google Scholar]
- Hughes, D.P.; Thomas, D.G.; Giordano, T.J.; Baker, L.H.; McDonagh, K.T. Cell surface expression of epidermal growth factor receptor and Her-2 with nuclear expression of Her-4 in primary osteosarcoma. Cancer Res 2004, 64, 2047–2053. [Google Scholar]
- Cottoni, F.; Ceccarelli, S.; Masala, M.V.; Montesu, M.A.; Satta, R.; Pirodda, C.; Rotolo, S.; Frati, L.; Marchese, C.; Angeloni, A. Overexpression of the fibroblast growth factor receptor 2-IIIc in Kaposi’s sarcoma. J. Dermatol. Sci 2009, 53, 65–68. [Google Scholar]
- Taylor, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Invest 2009, 119, 3395–3407. [Google Scholar]
- Ribatti, D.; Vacca, A.; Rusnati, M.; Presta, M. The discovery of basic fibroblast growth factor/fibroblast growth factor-2 and its role in haematological malignancies. Cytokine Growth Factor Rev 2007, 18, 327–334. [Google Scholar]
- Chesi, M.; Bergsagel, P.L.; Kuehl, W.M. The enigma of ectopic expression of FGFR3 in multiple myeloma: A critical initiating event or just a target for mutational activation during tumor progression. Curr. Opin. Hematol 2002, 9, 288–293. [Google Scholar]
- Jose, S.; Gotway, G.; Garcia, R.; Vaziri, A.; Tirado, C.A. 8p11–12 FGFR1 rearrangements in hematological malignancies: Review of the literature. J. Assoc. Genet. Technol 2010, 36, 203–208. [Google Scholar]
- Ronchetti, D.; Greco, A.; Compasso, S.; Colombo, G.; Dell’Era, P.; Otsuki, T.; Lombardi, L.; Neri, A. Deregulated FGFR3 mutants in multiple myeloma cell lines with t(4;14): Comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene 2001, 20, 3553–3562. [Google Scholar]
- Buhring, H.J.; Sures, I.; Jallal, B.; Weiss, F.U.; Busch, F.W.; Ludwig, W.D.; Handgretinger, R.; Waller, H.D.; Ullrich, A. The receptor tyrosine kinase p185HER2 is expressed on a subset of B-lymphoid blasts from patients with acute lymphoblastic leukemia and chronic myelogenous leukemia. Blood 1995, 86, 1916–1923. [Google Scholar]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol 2009, 21, 177–184. [Google Scholar]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol 1997, 8, 1197–1206. [Google Scholar]
- Hynes, N.E.; Stern, D.F. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim. Biophys. Acta 1994, 1198, 165–184. [Google Scholar]
- Baselga, J.; Perez, E.A.; Pienkowski, T.; Bell, R. Adjuvant trastuzumab: A milestone in the treatment of HER-2-positive early breast cancer. Oncologist 2006, 11, S4–S12. [Google Scholar]
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol 2006, 33, 369–385. [Google Scholar]
- Karamouzis, M.V.; Konstantinopoulos, P.A.; Papavassiliou, A.G. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol 2009, 10, 709–717. [Google Scholar]
- Ding, S.; Merkulova-Rainon, T.; Han, Z.C.; Tobelem, G. HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood 2003, 101, 4816–4822. [Google Scholar]
- Michieli, P.; Mazzone, M.; Basilico, C.; Cavassa, S.; Sottile, A.; Naldini, L.; Comoglio, P.M. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell 2004, 6, 61–73. [Google Scholar]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; van de Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol 2003, 4, 915–925. [Google Scholar]
- Ido, A.; Tsubouchi, H. Translational research to identify clinical applications of hepatocyte growth factor. Hepatol. Res 2009, 39, 739–747. [Google Scholar]
- Ponzo, M.G.; Lesurf, R.; Petkiewicz, S.; O’Malley, F.P.; Pinnaduwage, D.; Andrulis, I.L.; Bull, S.B.; Chughtai, N.; Zuo, D.; Souleimanova, M.; et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 12903–12908. [Google Scholar]
- Pacher, M.; Seewald, M.J.; Mikula, M.; Oehler, S.; Mogg, M.; Vinatzer, U.; Eger, A.; Schweifer, N.; Varecka, R.; Sommergruber, W.; et al. Impact of constitutive IGF1/IGF2 stimulation on the transcriptional program of human breast cancer cells. Carcinogenesis 2007, 28, 49–59. [Google Scholar]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar]
- Yee, D. The insulin-like growth factor system as a treatment target in breast cancer. Semin. Oncol 2002, 29, 86–95. [Google Scholar]
- Roberts, C.T., Jr. Control of insulin-like growth factor (IGF) action by regulation of IGF-I receptor expression. Endocr. J 1996, 43, S49–S55. [Google Scholar]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst 2001, 93, 1852–1857. [Google Scholar]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med 1995, 1, 27–31. [Google Scholar]
- Folkman, J. Tumor angiogenesis. Adv. Cancer Res 1985, 43, 175–203. [Google Scholar]
- Distler, J.H.; Hirth, A.; Kurowska-Stolarska, M.; Gay, R.E.; Gay, S.; Distler, O. Angiogenic and angiostatic factors in the molecular control of angiogenesis. Q. J. Nucl. Med 2003, 47, 149–161. [Google Scholar]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med 2003, 9, 669–676. [Google Scholar]
- Ortega, N.; Hutchings, H.; Plouet, J. Signal relays in the VEGF system. Front. Biosci 1999, 4, D141–D152. [Google Scholar]
- Roy, H.; Bhardwaj, S.; Yla-Herttuala, S. Biology of vascular endothelial growth factors. FEBS Lett 2006, 580, 2879–2887. [Google Scholar]
- Otrock, Z.K.; Makarem, J.A.; Shamseddine, A.I. Vascular endothelial growth factor family of ligands and receptors: Review. Blood Cells Mol. Dis 2007, 38, 258–268. [Google Scholar]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 2007, 19, 2003–2012. [Google Scholar]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar]
- Tortora, G.; Melisi, D.; Ciardiello, F. Angiogenesis: A target for cancer therapy. Curr. Pharm. Des 2004, 10, 11–26. [Google Scholar]
- Hurwitz, H. Integrating the anti-VEGF-A humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. Clin. Colorectal Cancer 2004, 4, S62–S68. [Google Scholar]
- Lu, D.; Shen, J.; Vil, M.D.; Zhang, H.; Jimenez, X.; Bohlen, P.; Witte, L.; Zhu, Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J. Biol. Chem 2003, 278, 43496–43507. [Google Scholar]
- Cooke, S.P.; Boxer, G.M.; Lawrence, L.; Pedley, R.B.; Spencer, D.I.; Begent, R.H.; Chester, K.A. A strategy for antitumor vascular therapy by targeting the vascular endothelial growth factor: Receptor complex. Cancer Res 2001, 61, 3653–3659. [Google Scholar]
- Vitaliti, A.; Wittmer, M.; Steiner, R.; Wyder, L.; Neri, D.; Klemenz, R. Inhibition of tumor angiogenesis by a single-chain antibody directed against vascular endothelial growth factor. Cancer Res 2000, 60, 4311–4314. [Google Scholar]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol 2001, 2, reviews3005:1–reviews3005:12. [Google Scholar]
- Basilico, C.; Moscatelli, D. The FGF family of growth factors and oncogenes. Adv. Cancer Res 1992, 59, 115–165. [Google Scholar]
- Folkman, J.; Shing, Y. Control of angiogenesis by heparin and other sulfated polysaccharides. Adv. Exp. Med. Biol 1992, 313, 355–364. [Google Scholar]
- Presta, M.; Moscatelli, D.; Joseph-Silverstein, J.; Rifkin, D.B. Purification from a human hepatoma cell line of a basic fibroblast growth factor-like molecule that stimulates capillary endothelial cell plasminogen activator production, DNA synthesis, and migration. Mol. Cell. Biol 1986, 6, 4060–4066. [Google Scholar]
- Javerzat, S.; Auguste, P.; Bikfalvi, A. The role of fibroblast growth factors in vascular development. Trends Mol. Med 2002, 8, 483–489. [Google Scholar]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res 1993, 60, 1–41. [Google Scholar]
- Giri, D.; Ropiquet, F.; Ittmann, M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin. Cancer Res 1999, 5, 1063–1071. [Google Scholar]
- Antoine, M.; Wirz, W.; Tag, C.G.; Mavituna, M.; Emans, N.; Korff, T.; Stoldt, V.; Gressner, A.M.; Kiefer, P. Expression pattern of fibroblast growth factors (FGFs), their receptors and antagonists in primary endothelial cells and vascular smooth muscle cells. Growth Factors 2005, 23, 87–95. [Google Scholar]
- Ronca, R.; Benzoni, P.; Leali, D.; Urbinati, C.; Belleri, M.; Corsini, M.; Alessi, P.; Coltrini, D.; Calza, S.; Presta, M.; et al. Antiangiogenic activity of a neutralizing human single-chain antibody fragment against fibroblast growth factor receptor 1. Mol. Cancer Ther 2010, 9, 3244–3253. [Google Scholar]
- Yayon, A.; Aviezer, D.; Safran, M.; Gross, J.L.; Heldman, Y.; Cabilly, S.; Givol, D.; Katchalski-Katzir, E. Isolation of peptides that inhibit binding of basic fibroblast growth factor to its receptor from a random phage-epitope library. Proc. Natl. Acad. Sci. USA 1993, 90, 10643–10647. [Google Scholar]
- Cardo-Vila, M.; Giordano, R.J.; Sidman, R.L.; Bronk, L.F.; Fan, Z.; Mendelsohn, J.; Arap, W.; Pasqualini, R. From combinatorial peptide selection to drug prototype (II): Targeting the epidermal growth factor receptor pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 5118–5123. [Google Scholar]
- Kumar, S.R.; Quinn, T.P.; Deutscher, S.L. Evaluation of an 111In-radiolabeled peptide as a targeting and imaging agent for ErbB-2 receptor expressing breast carcinomas. Clin. Cancer Res 2007, 13, 6070–6079. [Google Scholar]
- Hetian, L.; Ping, A.; Shumei, S.; Xiaoying, L.; Luowen, H.; Jian, W.; Lin, M.; Meisheng, L.; Junshan, Y.; Chengchao, S. A novel peptide isolated from a phage display library inhibits tumor growth and metastasis by blocking the binding of vascular endothelial growth factor to its kinase domain receptor. J. Biol. Chem 2002, 277, 43137–43142. [Google Scholar]
- Rastelli, L.; Valentino, M.L.; Minderman, M.C.; Landin, J.; Malyankar, U.M.; Lescoe, M.K.; Kitson, R.; Brunson, K.; Souan, L.; Forenza, S.; et al. A KDR-binding peptide (ST100,059) can block angiogenesis, melanoma tumor growth and metastasis in vitro and in vivo. Int. J. Oncol 2011, 39, 401–408. [Google Scholar]
- Fairbrother, W.J.; Christinger, H.W.; Cochran, A.G.; Fuh, G.; Keenan, C.J.; Quan, C.; Shriver, S.K.; Tom, J.Y.; Wells, J.A.; Cunningham, B.C. Novel peptides selected to bind vascular endothelial growth factor target the receptor-binding site. Biochemistry 1998, 37, 17754–17764. [Google Scholar]
- Erdag, B.; Balcioglu, K.B.; Kumbasar, A.; Celikbicak, O.; Zeder-Lutz, G.; Altschuh, D.; Salih, B.; Baysal, K. Novel short peptides isolated from phage display library inhibit vascular endothelial growth factor activity. Mol. Biotechnol 2007, 35, 51–63. [Google Scholar]
- Binetruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouet, J.; Derbin, C.; Perret, G.; et al. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J 2000, 19, 1525–1533. [Google Scholar]
- Nilsson, F.; Tarli, L.; Viti, F.; Neri, D. The use of phage display for the development of tumour targeting agents. Adv. Drug Deliv. Rev 2000, 43, 165–196. [Google Scholar]
- Yoo, M.K.; Kang, S.K.; Choi, J.H.; Park, I.K.; Na, H.S.; Lee, H.C.; Kim, E.B.; Lee, N.K.; Nah, J.W.; Choi, Y.J.; et al. Targeted delivery of chitosan nanoparticles to Peyer’s patch using M cell-homing peptide selected by phage display technique. Biomaterials 2010, 31, 7738–7747. [Google Scholar]
- Laakkonen, P.; Akerman, M.E.; Biliran, H.; Yang, M.; Ferrer, F.; Karpanen, T.; Hoffman, R.M.; Ruoslahti, E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 9381–9386. [Google Scholar]
- Zhu, Z.; Rockwell, P.; Lu, D.; Kotanides, H.; Pytowski, B.; Hicklin, D.J.; Bohlen, P.; Witte, L. Inhibition of vascular endothelial growth factor-induced receptor activation with anti-kinase insert domain-containing receptor single-chain antibodies from a phage display library. Cancer Res 1998, 58, 3209–3214. [Google Scholar]
- Erdag, B.; Balcioglu, B.K.; Bahadir, A.O.; Serhatli, M.; Kacar, O.; Bahar, A.; Seker, U.O.; Akgun, E.; Ozkan, A.; Kilic, T.; et al. Identification of novel neutralizing single-chain antibodies against vascular endothelial growth factor receptor 2. Biotechnol. Appl. Biochem 2011, 58, 412–422. [Google Scholar]
- Veggiani, G.; Ossolengo, G.; Aliprandi, M.; Cavallaro, U.; de Marco, A. Single-domain antibodies that compete with the natural ligand fibroblast growth factor block the internalization of the fibroblast growth factor receptor 1. Biochem. Biophys. Res. Commun 2011, 408, 692–696. [Google Scholar]
- Dong, J.; Sereno, A.; Aivazian, D.; Langley, E.; Miller, B.R.; Snyder, W.B.; Chan, E.; Cantele, M.; Morena, R.; Joseph, I.B.; et al. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. MAbs 2011, 3, 273–288. [Google Scholar]
- Lu, R.M.; Chang, Y.L.; Chen, M.S.; Wu, H.C. Single chain anti-c-Met antibody conjugated nanoparticles for in vivo tumor-targeted imaging and drug delivery. Biomaterials 2011, 32, 3265–3274. [Google Scholar]
- Lee, J.W.; Stone, R.L.; Lee, S.J.; Nam, E.J.; Roh, J.W.; Nick, A.M.; Han, H.D.; Shahzad, M.M.; Kim, H.S.; Mangala, L.S.; et al. EphA2 targeted chemotherapy using an antibody drug conjugate in endometrial carcinoma. Clin. Cancer Res 2010, 16, 2562–2570. [Google Scholar]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res 2011, 17, 6437–6447. [Google Scholar]
- Moldenhauer, G.; Salnikov, A.V.; Luttgau, S.; Herr, I.; Anderl, J.; Faulstich, H. Therapeutic Potential of Amanitin-Conjugated Anti-Epithelial Cell Adhesion Molecule Monoclonal Antibody Against Pancreatic Carcinoma. J. Natl. Cancer Inst 2012. [Google Scholar] [CrossRef]
- Green, L.L. Antibody engineering via genetic engineering of the mouse: XenoMouse strains are a vehicle for the facile generation of therapeutic human monoclonal antibodies. J. Immunol. Methods 1999, 231, 11–23. [Google Scholar]
- Lonberg, N. Fully human antibodies from transgenic mouse and phage display platforms. Curr. Opin. Immunol 2008, 20, 450–459. [Google Scholar]
- Louis, E.; Lofberg, R.; Reinisch, W.; Camez, A.; Yang, M.; Pollack, P.F.; Chen, N.; Chao, J.; Mulani, P.M. Adalimumab improves patient-reported outcomes and reduces indirect costs in patients with moderate to severe Crohn;s disease: Results from the CARE trial. J. Crohns Colitis 2012. [Google Scholar] [CrossRef]
- Yang, H.; Epstein, D.; Bojke, L.; Craig, D.; Light, K.; Bruce, I.; Sculpher, M.; Woolacott, N. Golimumab for the treatment of psoriatic arthritis. Health Technol. Assess 2011, 15, S87–S95. [Google Scholar]
- Ballestrero, A.; Garuti, A.; Cirmena, G.; Rocco, I.; Palermo, C.; Nencioni, A.; Scabini, S.; Zoppoli, G.; Parodi, S.; Patrone, F. Patient-tailored treatments with anti-EGFR monoclonal antibodies in advanced colorectal cancer: KRAS and beyond. Curr. Cancer Drug Targets 2012, 12, 316–328. [Google Scholar]
- Curran, M.P. Canakinumab: In patients with cryopyrin-associated periodic syndromes. BioDrugs 2012, 26, 53–59. [Google Scholar]
- Winchester, D.; Callis Duffin, K.; Hansen, C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic discoid lupus. Lupus 2012. [Google Scholar] [CrossRef]
- Hoyle, M.; Crathorne, L.; Garside, R.; Hyde, C. Ofatumumab for the treatment of chronic lymphocytic leukaemia in patients who are refractory to fludarabine and alemtuzumab: A critique of the submission from GSK. Health Technol. Assess 2011, 15, S61–S67. [Google Scholar]
- Waugh, N.; Royle, P.; Scotland, G.; Henderson, R.; Hollick, R.; McNamee, P. Denosumab for the prevention of osteoporotic fractures in postmenopausal women. Health Technol. Assess 2011, 15, S51–S59. [Google Scholar]
- Boyce, E.G.; Fusco, B.E. Belimumab: Review of use in systemic lupus erythematosus. Clin. Ther 2012. [Google Scholar] [CrossRef]
- Margolin, K.; Ernstoff, M.S.; Hamid, O.; Lawrence, D.; McDermott, D.; Puzanov, I.; Wolchok, J.D.; Clark, J.I.; Sznol, M.; Logan, T.F.; et al. Ipilimumab in patients with melanoma and brain metastases: An open-label, phase 2 trial. Lancet Oncol 2012, ((12)), 70090–6. [Google Scholar] [CrossRef]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab: First global approval. Drugs 2011, 71, 1093–1104. [Google Scholar]
- Migone, T.S.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med 2009, 361, 135–144. [Google Scholar]
- Thie, H.; Meyer, T.; Schirrmann, T.; Hust, M.; Dubel, S. Phage display derived therapeutic antibodies. Curr. Pharm. Biotechnol 2008, 9, 439–446. [Google Scholar]
- Rowinsky, E.K.; Youssoufian, H.; Tonra, J.R.; Solomon, P.; Burtrum, D.; Ludwig, D.L. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin. Cancer Res 2007, 13, 5549s–5555s. [Google Scholar]
- Erinjeri, J.P.; Deodhar, A.; Thornton, R.H.; Allen, P.J.; Getrajdman, G.I.; Brown, K.T.; Sofocleous, C.T.; Reidy, D.L. Resolution of hepatic encephalopathy following hepatic artery embolization in a patient with well-differentiated neuroendocrine tumor metastatic to the liver. Cardiovasc. Intervent. Radiol 2010, 33, 610–614. [Google Scholar]
- Beltran, P.J.; Mitchell, P.; Chung, Y.A.; Cajulis, E.; Lu, J.; Belmontes, B.; Ho, J.; Tsai, M.M.; Zhu, M.; Vonderfecht, S.; et al. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol. Cancer Ther 2009, 8, 1095–1105. [Google Scholar]
- Tolcher, A.W.; Sarantopoulos, J.; Patnaik, A.; Papadopoulos, K.; Lin, C.C.; Rodon, J.; Murphy, B.; Roth, B.; McCaffery, I.; Gorski, K.S.; et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J. Clin. Oncol 2009, 27, 5800–5807. [Google Scholar]
- Beltran, P.J.; Chung, Y.A.; Moody, G.; Mitchell, P.; Cajulis, E.; Vonderfecht, S.; Kendall, R.; Radinsky, R.; Calzone, F.J. Efficacy of ganitumab (AMG 479), alone and in combination with rapamycin, in Ewing’s and osteogenic sarcoma models. J. Pharmacol. Exp. Ther 2011, 337, 644–654. [Google Scholar]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomaki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellstrom, M.; Schomber, T.; Peltonen, R.; et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar]
- Kuenen, B.; Witteveen, P.O.; Ruijter, R.; Giaccone, G.; Dontabhaktuni, A.; Fox, F.; Katz, T.; Youssoufian, H.; Zhu, J.; Rowinsky, E.K.; et al. A phase I pharmacologic study of necitumumab (IMC-11F8), a fully human IgG1 monoclonal antibody directed against EGFR in patients with advanced solid malignancies. Clin. Cancer Res 2010, 16, 1915–1923. [Google Scholar]
- Lu, D.; Jimenez, X.; Zhang, H.; Bohlen, P.; Witte, L.; Zhu, Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int. J. Cancer 2002, 97, 393–399. [Google Scholar]
- Karkkainen, M.J.; Jussila, L.; Ferrell, R.E.; Finegold, D.N.; Alitalo, K. Molecular regulation of lymphangiogenesis and targets for tissue oedema. Trends Mol. Med 2001, 7, 18–22. [Google Scholar]
- Achen, M.G.; Stacker, S.A. Molecular control of lymphatic metastasis. Ann. NY Acad. Sci 2008, 1131, 225–234. [Google Scholar]
- Grau, S.J.; Trillsch, F.; Herms, J.; Thon, N.; Nelson, P.J.; Tonn, J.C.; Goldbrunner, R. Expression of VEGFR3 in glioma endothelium correlates with tumor grade. J. Neurooncol 2007, 82, 141–150. [Google Scholar]
- Hajrasouliha, A.R.; Funaki, T.; Sadrai, Z.; Hattori, T.; Chauhan, S.K.; Dana, R. Vascular endothelial growth factor-C promotes alloimmunity by amplifying antigen-presenting cell maturation and lymphangiogenesis. Invest. Ophthalmol. Vis. Sci 2012, 53, 1244–1250. [Google Scholar]
- Grutter, C.; Wilkinson, T.; Turner, R.; Podichetty, S.; Finch, D.; McCourt, M.; Loning, S.; Jermutus, L.; Grutter, M.G. A cytokine-neutralizing antibody as a structural mimetic of 2 receptor interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 20251–20256. [Google Scholar]
- Ronca, R.; Sozzani, S.; Presta, M.; Alessi, P. Delivering cytokines at tumor site: The immunocytokine-conjugated anti-EDB-fibronectin antibody case. Immunobiology 2009, 214, 800–810. [Google Scholar]
- Kurtz, J.E.; Dufour, P. Adecatumumab: An anti-EpCAM monoclonal antibody, from the bench to the bedside. Expert Opin. Biol. Ther 2010, 10, 951–958. [Google Scholar]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J. Control. Release 2012. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tumor type | Growth factor | RTK | References |
|---|---|---|---|
| Glioma/Glioblastoma | FGF2, VEGFC | EGFR | [22–25] |
| Head-neck squamous cell | Met, FGFR1, FGFR3, EGFR | [26–28] | |
| Esophageal and Gastric | FGF2 | Met | [29–31] |
| Liver/Hepatocarcinoma | Met, VEGR2 | [32,33] | |
| Colorectal | FGF2, VEGFA | Met, ErbB2, IGF1R | [34–38] |
| Lung | FGF2 | FGFR1, FGFR2, EGFR | [39–41] |
| Renal | FGF2 | Met, IGF1R, VEGFR2 | [32,42–44] |
| Prostate | FGF2, FGF8 | FGFR1, FGFR3, ErbB2, IGF1R | [45–48] |
| Bladder | FGFR3, ErbB2 | [49,50] | |
| Ovarian | FGFR2, ErbB2, IGF1R | [40,51,52] | |
| Breast | FGF8 | FGFR1, FGFR2, EGFR, ErbB2, IGF1R | [40,52–55] |
| Melanoma | FGFR1, FGFR2, VEGFR2 | [32,56] | |
| Pancreatic | FGFR1 | [57] | |
| Sarcomas | FGFR2, FGFR4, EGFR, IGF1R | [58–61] | |
| Hematological malignancies | FGF2 | Met, ErbB2, FGFR1, FGFR3 | [52,62–66] |
| Molecule | Company | Commercial name | Target | Ref |
|---|---|---|---|---|
| IMC-A12 | ImClone LLC | Cixutumumab | IGF1R | [140,141] |
| AMG-479 | Amgen | Ganitumab | IGF1R | [142–144] |
| IMC-11F8 | ImClone LLC | Necitumumab | EGFR | [139,145,146] |
| IMC-1121b | ImClone LLC | Ramucirumab | VEGFR2 | [32,147] |
| IMC-3C5 | ImClone LLC | - | VEGFR3 | [145,148–150] |
| VGX-100 | Circadian Technologies Ltd | Fresolimumab | VEGFC | [151] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ronca, R.; Benzoni, P.; De Luca, A.; Crescini, E.; Dell’Era, P. Phage Displayed Peptides/Antibodies Recognizing Growth Factors and Their Tyrosine Kinase Receptors as Tools for Anti-Cancer Therapeutics. Int. J. Mol. Sci. 2012, 13, 5254-5277. https://doi.org/10.3390/ijms13045254
Ronca R, Benzoni P, De Luca A, Crescini E, Dell’Era P. Phage Displayed Peptides/Antibodies Recognizing Growth Factors and Their Tyrosine Kinase Receptors as Tools for Anti-Cancer Therapeutics. International Journal of Molecular Sciences. 2012; 13(4):5254-5277. https://doi.org/10.3390/ijms13045254
Chicago/Turabian StyleRonca, Roberto, Patrizia Benzoni, Angela De Luca, Elisabetta Crescini, and Patrizia Dell’Era. 2012. "Phage Displayed Peptides/Antibodies Recognizing Growth Factors and Their Tyrosine Kinase Receptors as Tools for Anti-Cancer Therapeutics" International Journal of Molecular Sciences 13, no. 4: 5254-5277. https://doi.org/10.3390/ijms13045254
APA StyleRonca, R., Benzoni, P., De Luca, A., Crescini, E., & Dell’Era, P. (2012). Phage Displayed Peptides/Antibodies Recognizing Growth Factors and Their Tyrosine Kinase Receptors as Tools for Anti-Cancer Therapeutics. International Journal of Molecular Sciences, 13(4), 5254-5277. https://doi.org/10.3390/ijms13045254