1. Introduction
Meningioma cell
in vitro culture is the foundation of meningioma pathogenesis and drug treatment research [
1]. So far, a stable and efficient method for meningioma cell cultures has not been established [
2–
5]. As we know, pre-clinical studies of meningioma depends on the
in vitro and
in vivo tumor model,
in vivo models are also dependent on efficient cell models
in vitro. However, primary and subcultured human meningioma cells are difficult, compared with other tumor cells. Therefore, establishing an efficient meningioma cell model
in vitro becomes a necessary means for investigating the molecular basis of their growth and development, and subsequently studying meningioma pathogenesis and drug treatment. But the long-term subculture is difficult, a lot of meningioma research has been conducted mostly in primary cell-based cultures [
6–
8]. Although several strains have been established, except for malignant ones, most of the cell lines achieve immortalization under gene intervention, which is bound to change the characteristics of the original tumor cells [
9]. Therefore, the establishment of efficient meningioma cells in primary culture and subculture systems is important for the study of the biology and development of new drug treatments.
However, it is difficult to obtain a large number of primary meningioma cells and subculture cells, due to the following factors: (1) Meningioma tissue contains a high percentage of interstitial matrix/fiber, and it is difficult to digest meningiomas using a single enzyme or mechanical methods. And long enzymatic digestion or rigid mechanical separation result in cell disruption and fewer living cells. (2) Since the majority of meningiomas are benign, cells grow slowly surrounding the tissue and cannot spread far away into the tissue. (3) A system for the cultivation of meningioma cells has not been established, and it is not clear what kind of growth factors or hormones could promote the proliferation of meningioma cells.
In a previous study, our lab established a comprehensive digestion method, which consisted of a collagenase/dispase/DNase cocktail combined with four-step digestion. Hueng
et al. have demonstrated the existence of human meningioma cells showing stem-like features of sphere-forming ability, self-renewal, and CD133 marker [
10]. And it has been demonstrated that hypoxia could enhance the generation of embryonic stem cells and induced pluripotent stem cells [
11]. In addition, progesterone and androgen receptor-positive rates in meningioma tissues are about 60~90% and 65%, respectively [
12,
13]. A defined medium conditioned by a cell culture might contain some unknown factors, perhaps capable of stimulating its own proliferation [
14–
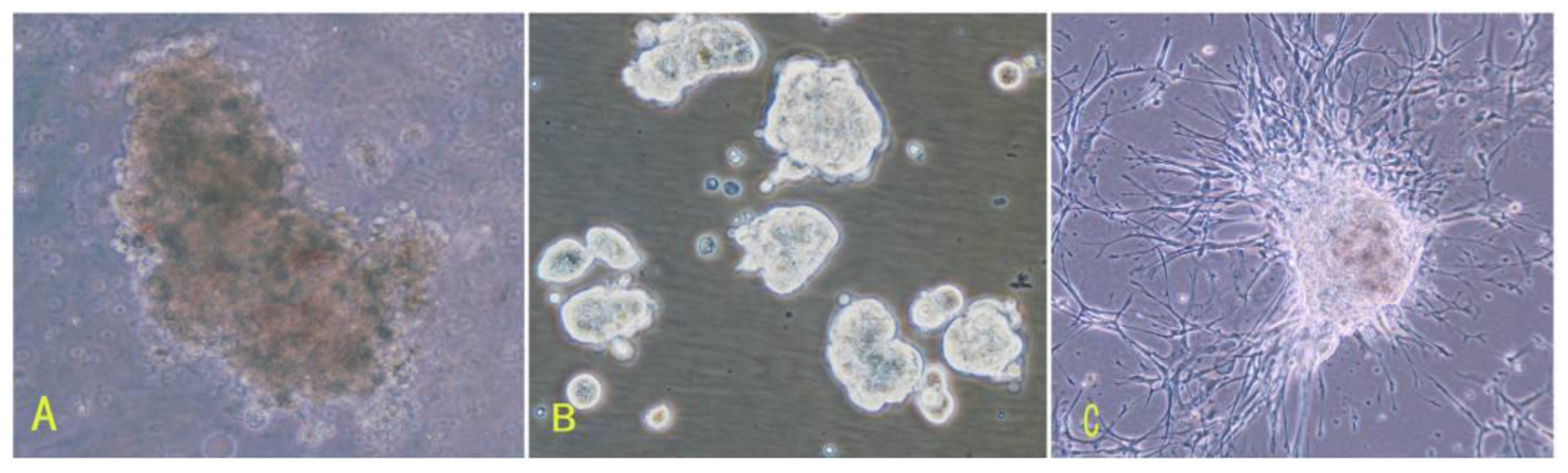
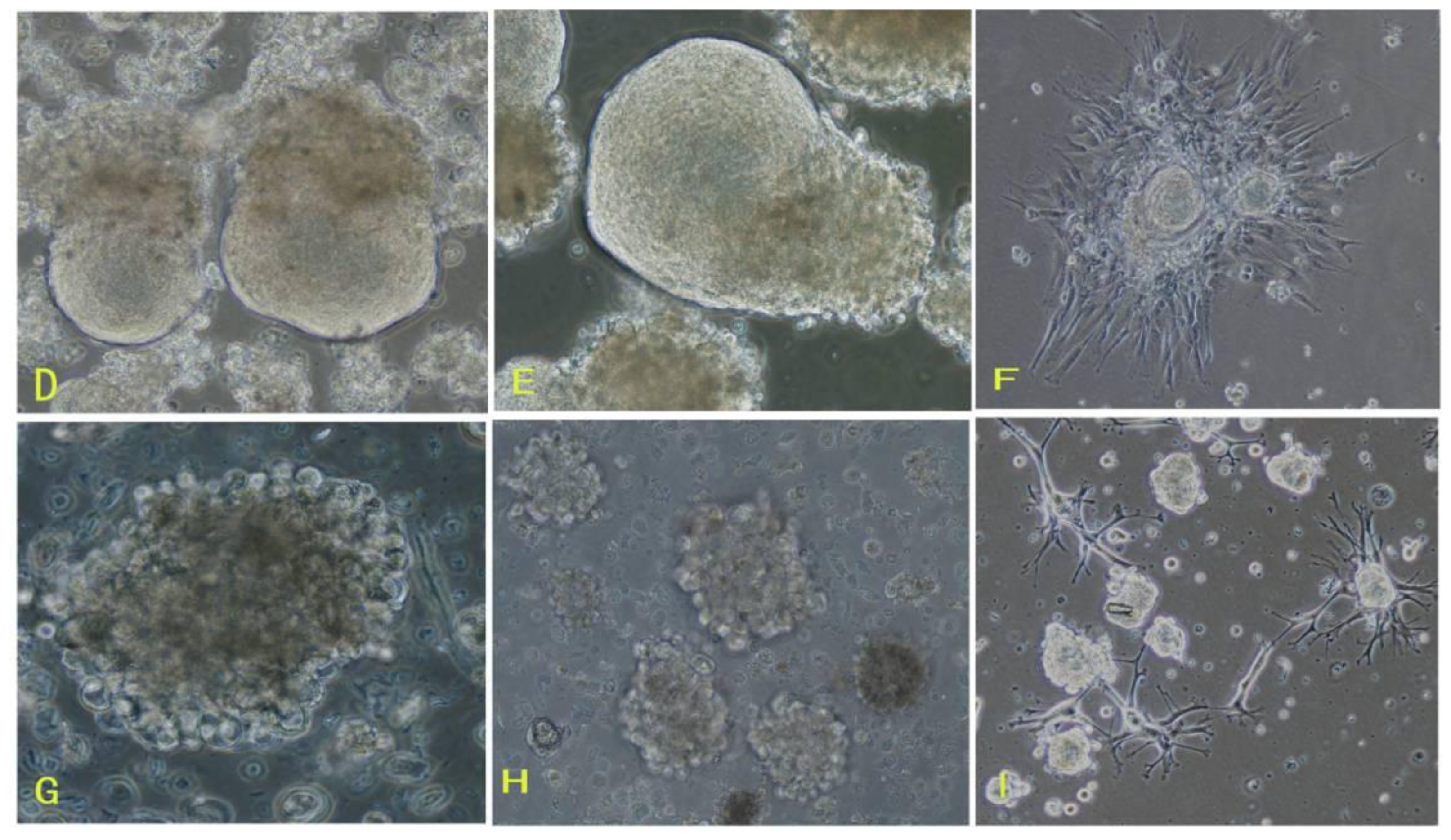
16]. To overcome those difficulties of meningioma cell culture mentioned above, in this study, we applied the comprehensive digestion method to digest meningioma tissues, then culture primary cells in the defined stem cell medium and hypoxia system. A large number of primary meningioma cells were obtained from six human meningioma tissues digested with our comprehensive digestion method. We found that these primary meningioma cells could proliferate and subculture over ten passages under sphere status in a conditioned medium and hypoxia system. Cells in sphere expressed not only epithelial membrane antigen and vimentin markers, but also CD133. Importantly, we found that the CD133 expression level was related with the cell proliferation rate. Our results suggest that stable and efficient culture systems for meningioma cells can be established under certain conditions.
3. Discussion
Meningiomas have the second highest incidence in original intracranial tumors [
17]. However, owing to the lack of appropriate model systems, investigations of their biology have been come to a halt. Since most meningiomas are benign, primary meningioma cells in culture are often found at senescence, resulting in the fact that studies on meningiomas had to depend on primary or early passage cells, except for those cell lines from aggressive variants of Meningiomas or immortalized cell lines. Our study showed that meningioma cells under defined conditions could be passaged over fifteen generations. Several factors below may contribute to the successful culture.
First, the comprehensive method of digestion used in this study contributed to obtaining living cells. To establish meningioma cell culture model systems, we developed a comprehensive digestion method, which consisted of three enzyme cocktail recipes and a four-step digestion method. By using the comprehensive method of digestion, we obtained a large number of living cells from each subtype of meningioma. These living cells would be easier to grow and propagate.
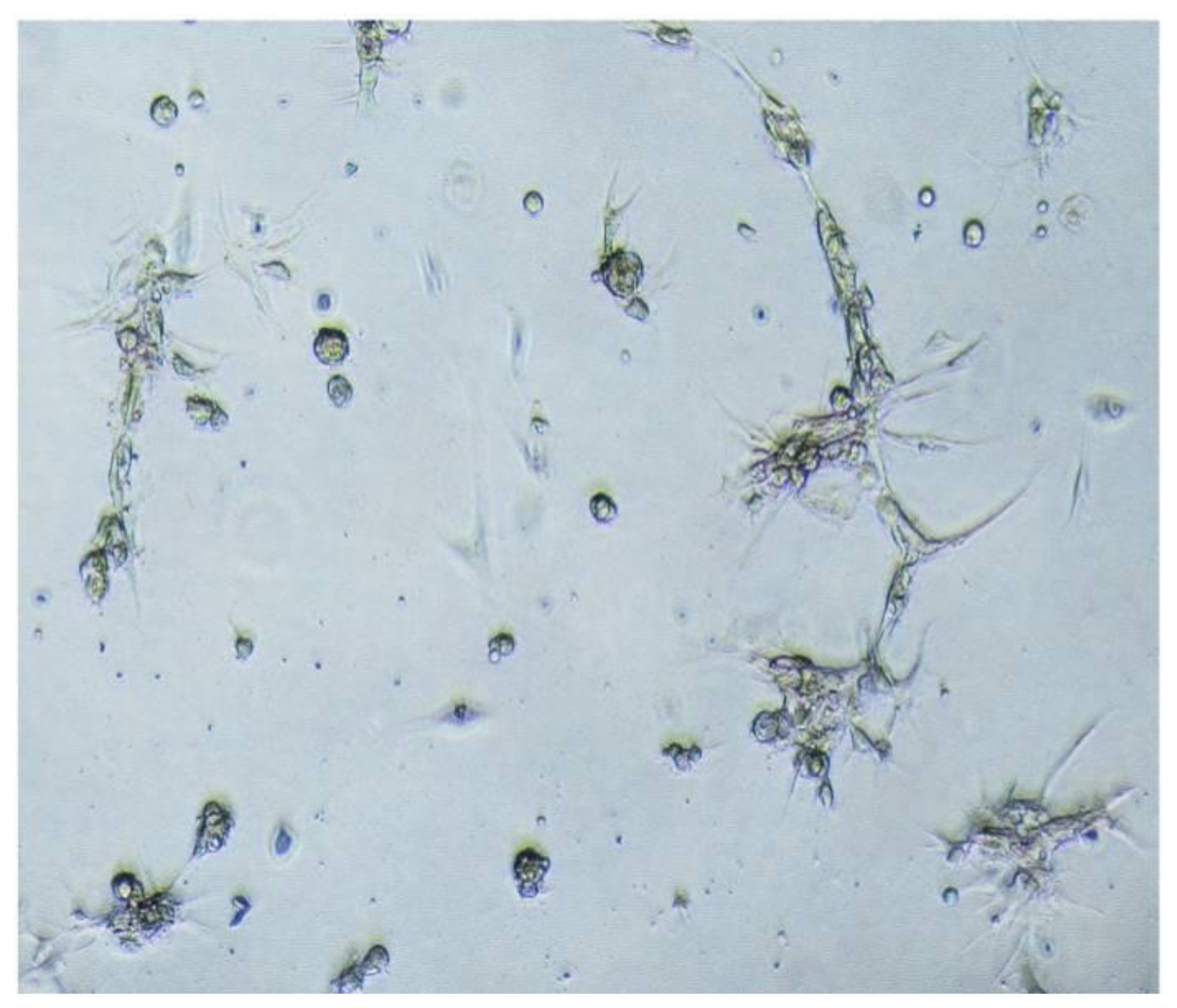
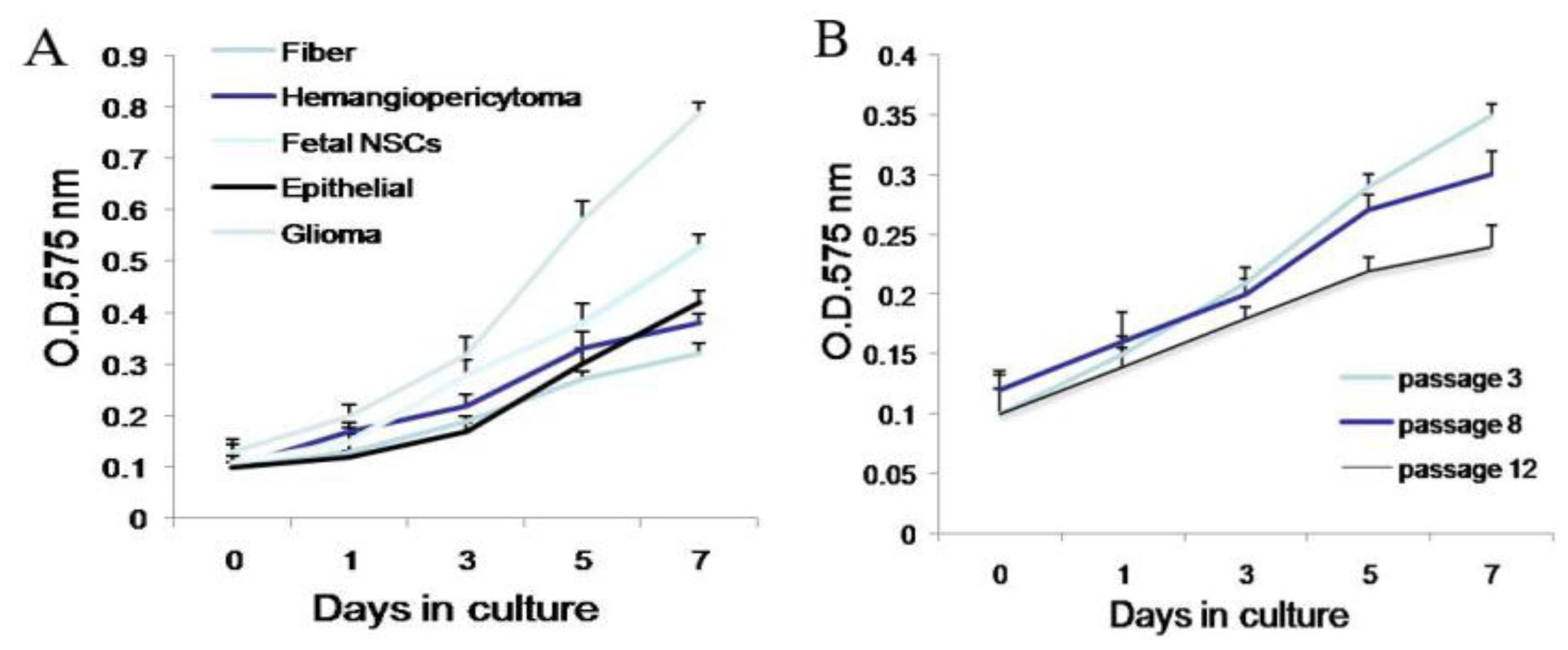
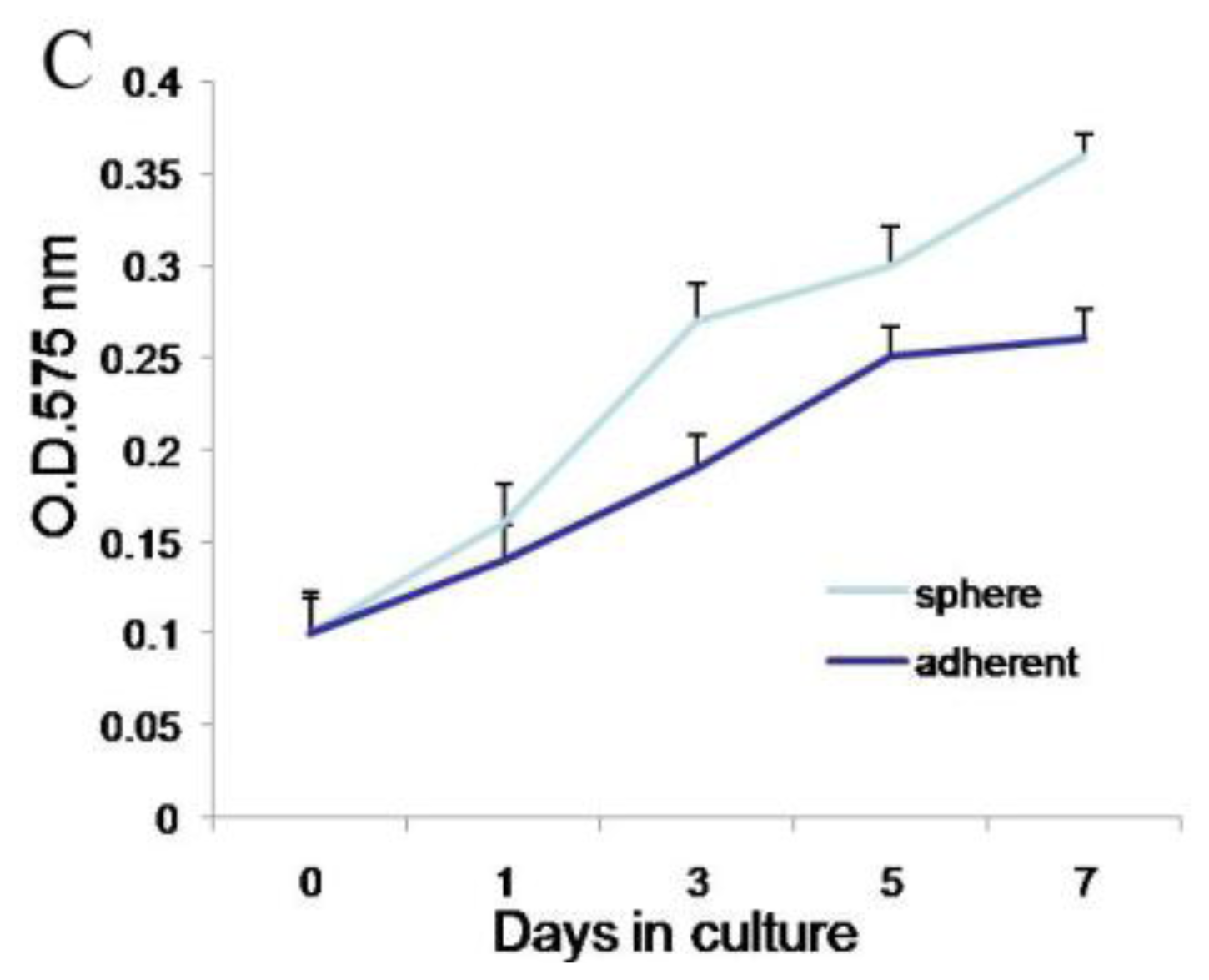
Second, meningioma cells under suspended spheres status might favor cell proliferation. Since meningioma cells in monolayer culture were react easily to contact inhibition and enter the quiescent and senescence states [
2–
5], we cultured meningioma cells under suspended spheres, thus avoiding contact inhibition, and so found that meningioma cells were not easy to enter the quiescent and senescence state. Our study showed that meningioma cells under suspended spheres could be passaged over ten generations. On the contrary, meningioma cells in adherent monolayer cultures were only passaged for four to six generations.
Third, there may be stem-like cells in meningiomas. The culture medium and environment in our study were appropriate for the growth and proliferation of stem cells. The culture medium in our study was conditional NSCs medium containing progesterone and androstenedione, and the culture system was a hypoxia system. The NSCs medium in our study is suitable for stem cells culture. Our results suggested that there would be stem-like cells in meningiomas, which is consistent with recent the identification of tumor stem-like cells as tumor-initiating cells in meningiomas [
10]. In addition, a lot of studies have proved that hypoxia could enhance the generation of embryonic stem cells and induce pluripotent stem cells [
11]. Therefore, we used NSCs medium as the basic medium for culturing meningioma cells in a hypoxic environment. In addition, so far, it is not clear what kind of growth factors can promote the proliferation of meningioma cells. However, defined medium conditioned by meningioma cells
per se might contain some factors, perhaps identical to a normal endogenous growth factor
in vivo, capable of stimulating the proliferation of meningioma cells [
14]. And the progesterone and androgen receptor-positive rate in meningioma tissues were relatively high [
12,
13], so progesterone and androstenedione in the culture medium might contribute to the growth and proliferation of meningioma cells.
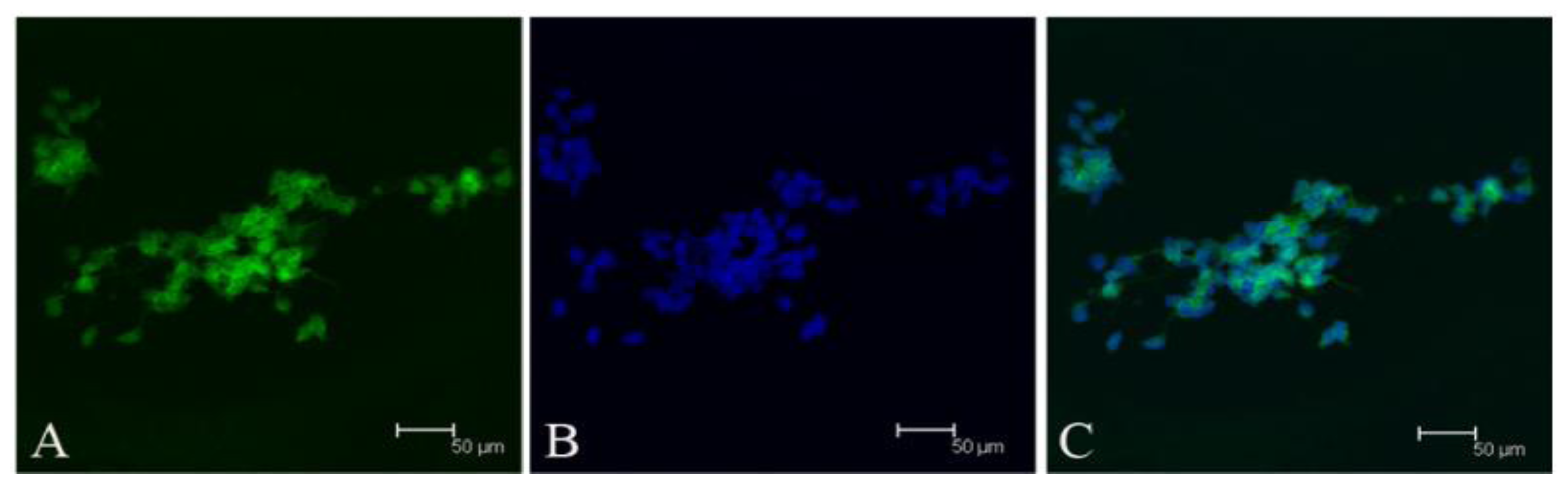
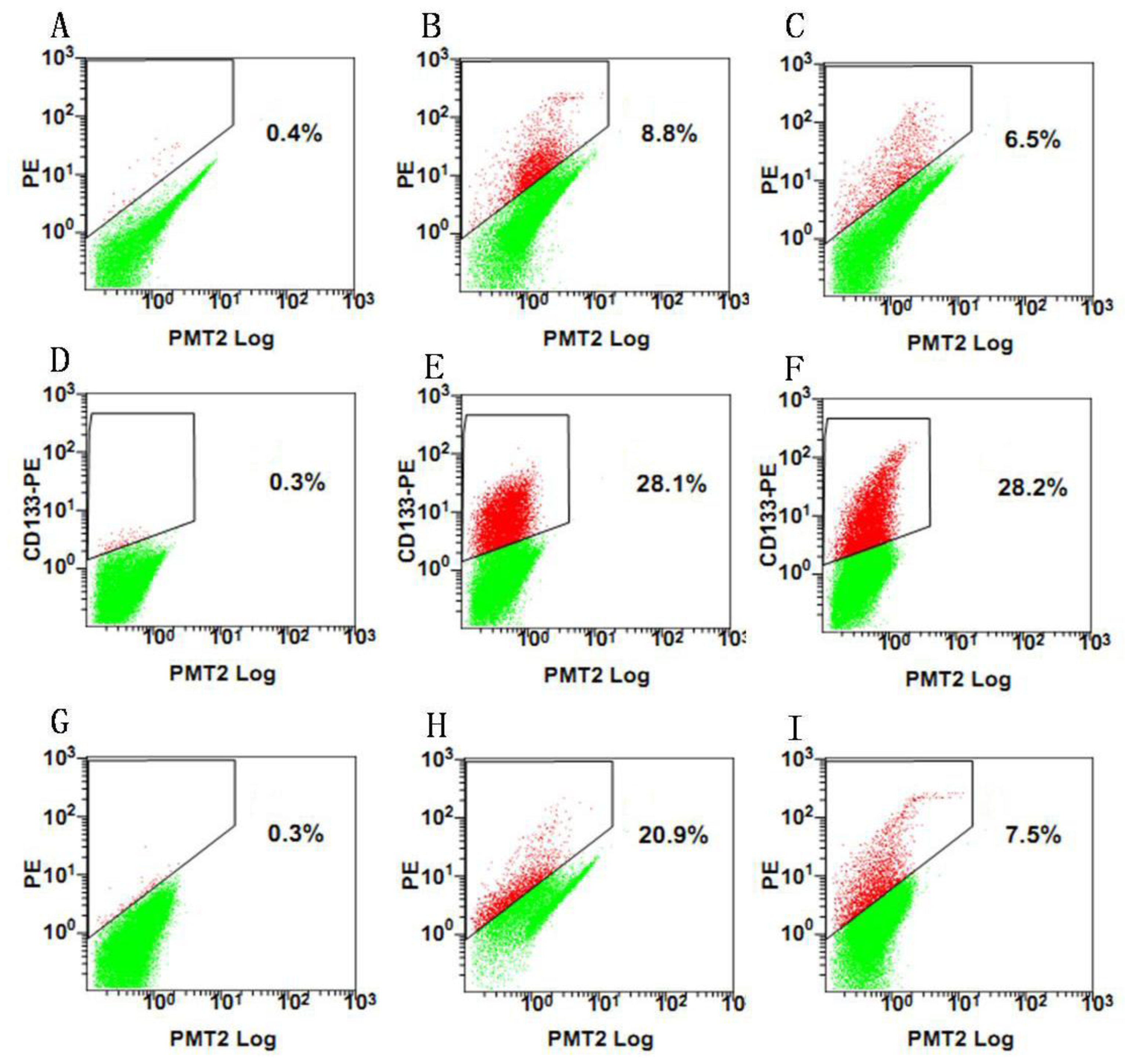
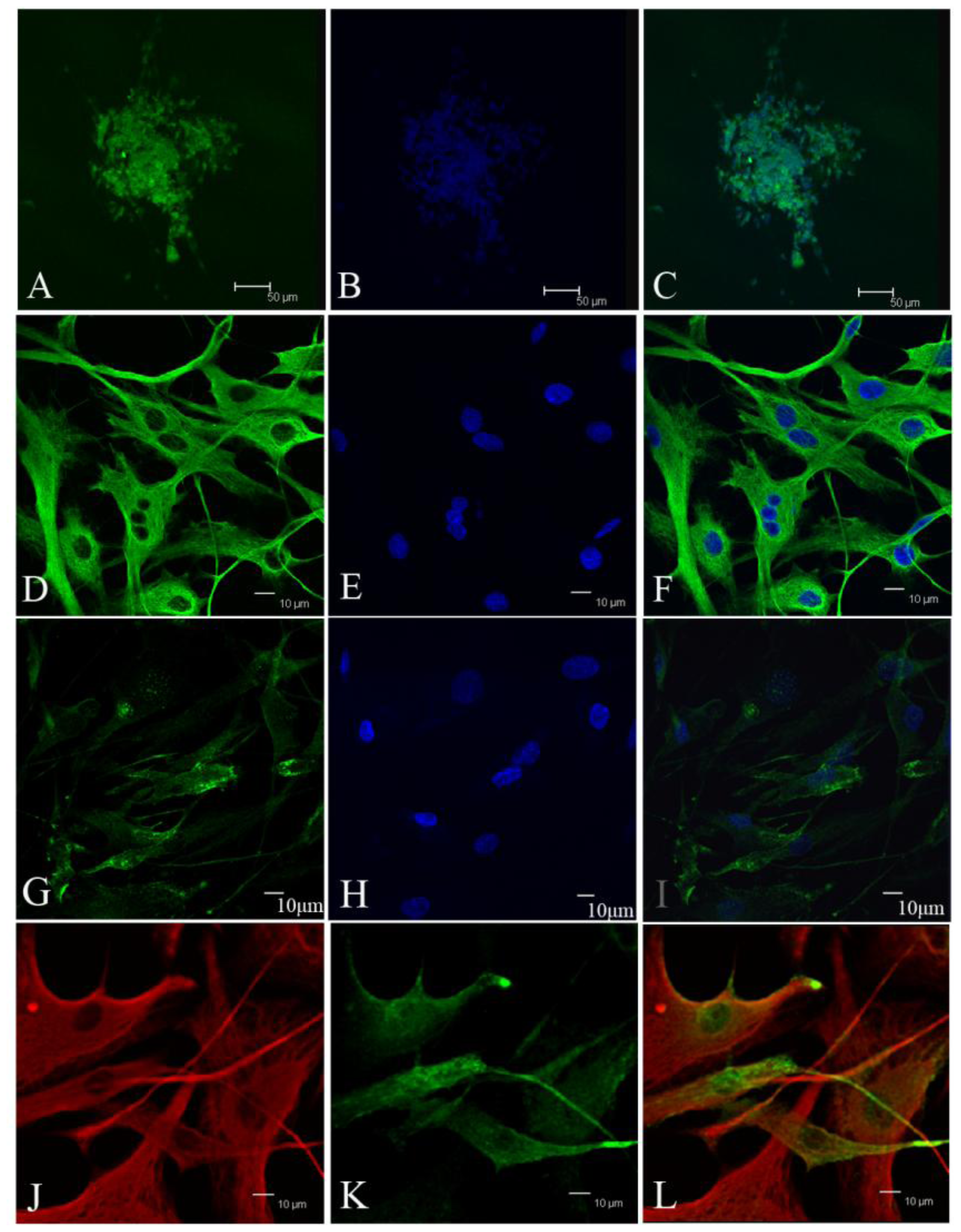
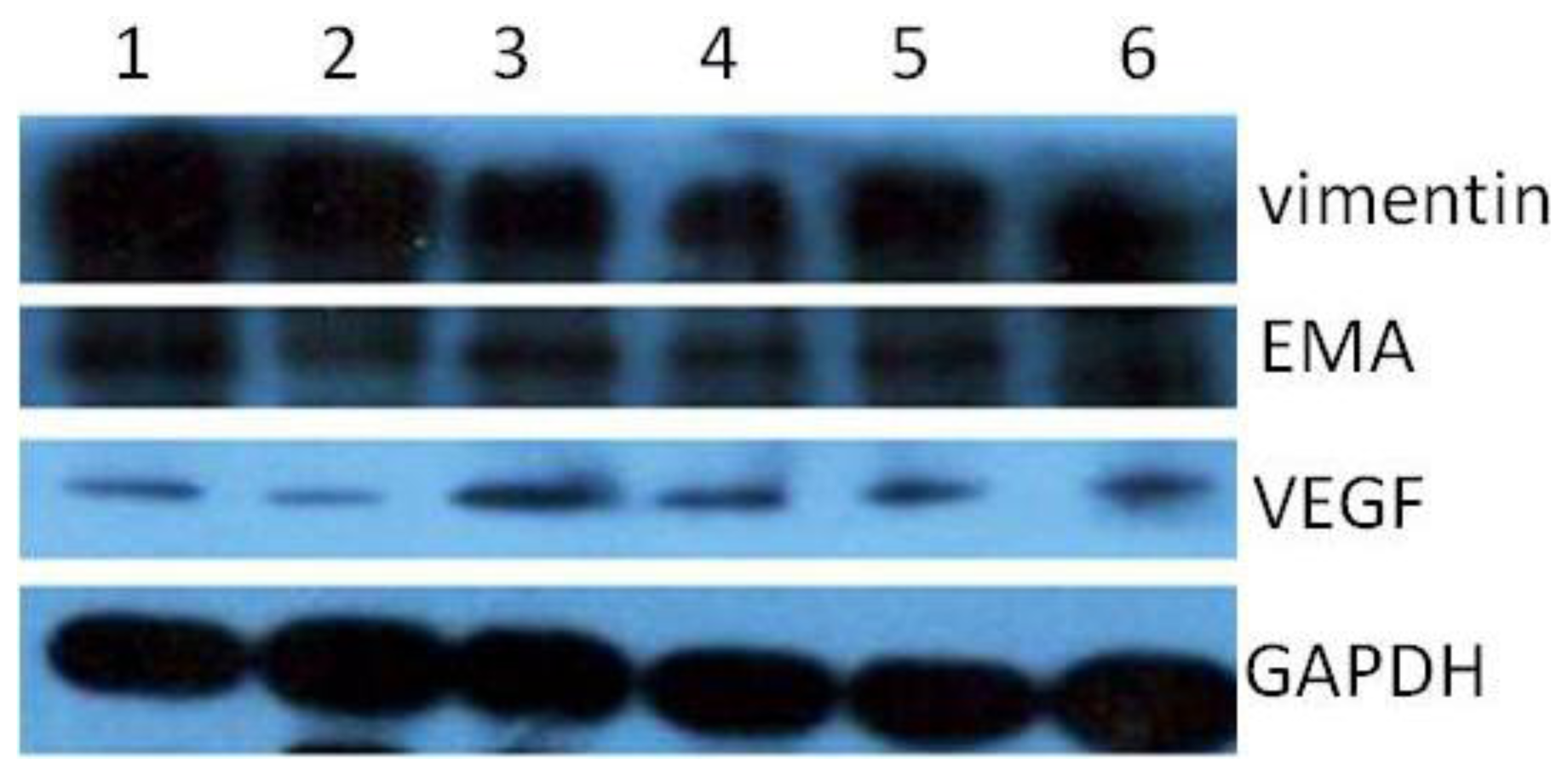
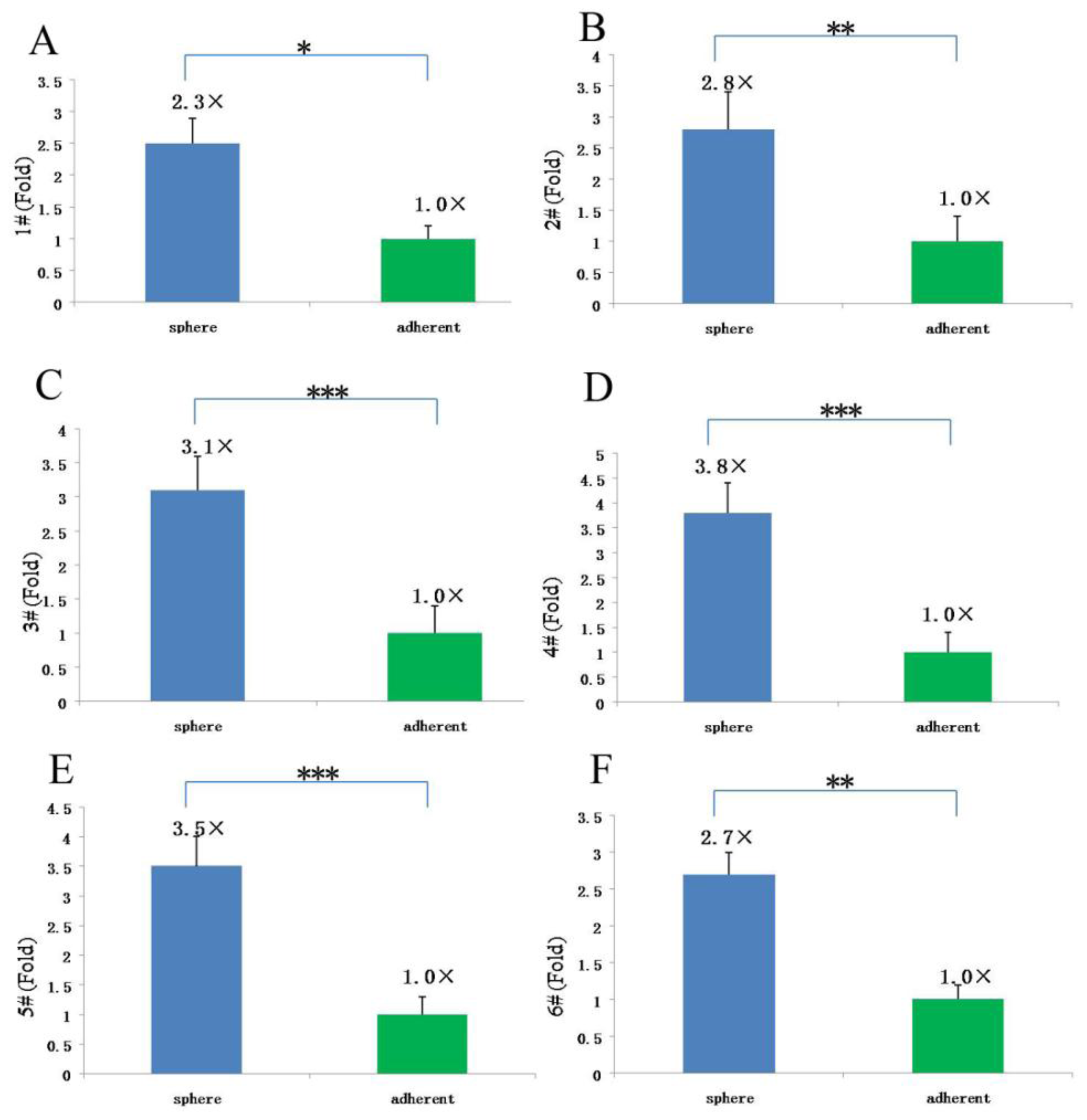
Since our results indicate that there may be stem-like cells in suspended sphere, we further investigated the expression level of CD133 of cells from different subtype tissues, different culture status, and different generations. For the same subtype of cells in different generations, the expression of CD133 of the earlier generation was obviously higher than that of the later generation. During the passage culture, we found a decreasing tendency of cell proliferation with increasing cell generation, suggesting that this decreasing tendency may be related with a decline of CD133 expression. Interestingly, cells from two epithelial subtypes could be passaged up to 15 generations, however, cells from fiber subtypes were only passaged up to 10 generations. Accordingly, CD133 expression in epithelial subtypes was higher than that in fiber subtypes, further suggesting there was a correlation between CD133 expression and cell proliferation rate. For the same subtype of cells under different status, we found that cells under suspension could be passaged for more generations, compared with those in adherent culture. Accordingly, the expression level of CD133 in cells under suspended spheres was significantly higher than that under adherence. Finally, single CD133+ cells rather than CD133− cells could form spheres. All of this indicated that CD133-positive cells might be responsible for efficient proliferation of human meningioma cells
During the passage culture, we found that some meningioma cells could form a vascular net. Immunocytochemistry detection demonstrated that there was VEGF expression in some of the cells. Relationships between stem cells and vascular cells suggested that these VEGF-positive cells might derive from CD133-positive cell transdifferentation [
18,
19].
In this study, we also transplanted meningioma cells into animal models to recapitulate the original tumor. However, we did not observe the tumor (data not shown). We speculated that stem cells in meningioma tissues might only have the characteristics of tissue stem cell-like cells. Therefore, meningioma cells in our study only passaged for limited generations, similar to adult neural stem cells.
In conclusion, our study demonstrated that meningioma cells could be amplicated and passaged under sphere status and conditional system without gene intervention, but the proliferative rate declined with cell generation and decreasing CD133 expression. In our latest study, we obtained a number of clinical specimens (including three fiber meningiomas, two epithelial meningiomas, two hemangiopericytom meningiomas). The growth characteristics and trends of meningioma cells from these specimens were similar to the corresponding subtypes of this study. Therefore, CD133-positve cells might be responsible for the proliferation, and decrease of CD133 level might be one of main causes for the decline of proliferative rate.
4. Experimental Section
4.1. Ethics
The informed consents from all participants involved in this study were obtained verbally. All procedures were approved by Human Research Subjects of Huashan Hospital, Fudan University, Shanghai, China.
4.2. Isolation of Meningioma Cells from Meningioma Tissues
Tumor samples from six human meningioma patients were removed by craniotomy, stored in a medium in a sterile test tube and transported to the lab at 4 °C. Tumor tissues were washed ten times with pre-cooled PBS, and cut into pieces. Tissue pieces were put into a 15 mL tube, washed in PBS and centrifuged for 5 min at 500 × g. Aspirate supernatant was digested in PBS-buffered collagenase/dispase cocktail, which consisted of 1 mg/mL collagenase, 2 mg/mL dispase, 70 U/mL DNase, 0.1% BSA. Digestion was performed for 4 × 20 min in a 37 °C rotator oven (four-step method). After the first 20 min, samples were gently triturated several times with 5 mL stripette (Corning Incerperated), and settled for 5 min. The digested top tissue mixture was transferred into a new tube to centrifuge and collect cells. Some fresh collagenase/dispase cocktail was added into the remaining tissue pieces and digest was continued for the second, third and forth 20 min. During each 20 min, the manipulation described for the first 20 min was repeated.
4.3. Primary Tumor Spheres Culture
Cells were seeded at a concentration of 5 × 105/mL into substrate-free tissue culture flasks and culture in a 3% O2, 5% CO2 atmosphere. The growth medium consisted of DMEM/F12 (phenol red-free) containing 50 units/mL penicillin–streptomycin, B27 (1:50), human recombinant FGF-2 and EGF (both at 20 ng/mL), 20 nM progesterone, 100 mM androstenedione and heparin (5 μg/mL).
4.4. Passaging Sphere Culture
Passaging was carried out using TrypleTM Express (Invitrogen, Carlsbad, CA, USA) and 70 U/mL DNase. When the majority of spheres were loose, 5 mL PBS were added and gently triturated, centrifuged and the cells collect at 300 g for 5 min. Cells were seeded in conditioned medium, which consisted of 1/2 of old medium and 1/2 of fresh medium. During passaging culture, 1/2 of the medium was changed every 3 days to keep it conditioned at all times.
4.5. Adherent Culture of Meningioma Cells
Spheres were triturated into single cell suspension, plated in T-25 flasks with a volume of 5 mL of conditional medium with 10% FBS. The growth medium consisted of DMEM/F12 containing 50 units/mL penicillin–streptomycin, B27 (1:50, Gibco, Grand Island, NY, USA), human recombinant FGF-2 and EGF (both at 20 ng/mL), 20 nM progesterone, 100 mM androstenedione, heparin (5 μg/mL), and 10% fetal bovine serum (FBS). The cells were passaged according to split ratios: 1:3 or 1:2.
4.6. Cell Proliferation Assays
Cells were plated in 96-well plates in 100 μL volumes of growth medium, at a density of 1000 cells/well. Cell proliferation assays were performed on days 0, 3, 5, and 7 post plating using the Cell Proliferation Kit I (MTT). Cells were incubated with MTT solution for approximately 4 h. The dye was quantified at 575 nm with an ELISA plate reader.
4.7. FACS Analysis
Tumor spheres were digested into single cells. The cell suspension was centrifuged at 300 g for 5 min. The supernatant was aspirated completely and the cells were pre-incubated with 20 μL of Affinity Purified Human FcγR-binding inhibitor per 100 μL Flow Cytometry Staining buffer (FCS buffer) for 20 min on ice prior to staining. Ten microliters of PE-labeled Anti-CD133 antibody (Miltenyi Biotec, Inc.) was added, mixed well and incubated for 30 min in the dark in a 4 °C refrigerator. Cells were washed by adding 2 mL of FCS buffer and centrifuged at 300 g, 4 °C for 5 min. This was repeated for a total of two washes, discarding supernatant between washes. The stained cells were resuspended in FCS buffer. Data was acquired on a FACSCalibur machine (BD Biosciences). After sorting, 1 CD133+ and CD133− were replated into each well to evaluate the proliferative capacity of CD133+ and CD133− cells.
4.8. Immunocytochemistry
Tumor sphere were partially digested into cells and loose spheres. Then these cells and loose spheres were replated on gelatin-coated glasses and cultured for 24 h. Fixed cells with 4% paraformaldehyde for 5 min at room temperature. After washing three times with PBS, the cells were treated with 0.25% Triton X-100, 10% normal donkey serum (Jackson), and 1% bovine serum albumin (BSA, Sigma) for 10 min at room temperature. Primary antibodies included CD133 (ab19898, abcam), vimentin (v6630, sigma), EMA (Dako, Carpinteria, CA, USA), Desmoplakin (ab109445, abcam), Nestin (Chemicon), bIII-tubulin (CB412, Chemicon), glial fibrillary acidic protein (Z0334, Dako). Secondary antibodies used were Alexa488-conjugated donkey anti-rabbit IgG (Invitrogen), Alexa555-conjugated donkey anti-mouse IgG (Invitrogen). The counterstain of nucleuses was 1 mg/mL Hoechst 33342 (Invitrogen).
4.9. qRT-PCR Analysis
The methods have been described previously [
15]. Total RNA was prepared from cells using the Absolutely RNA Nanoprep Kit (Stratagene,). The forward and reverse primers of CD133 were 5′-TCT CTA TGT GGT ACA GCC G-3′, 5′-TGA TCC GGG TTC TTA CCT-3′. Complementary DNA synthesis and quantitative analysis were performed with AffiniScriptTM Muti Temperature cDNA Synthesis Kit and Brilliant II Fast SYBR Green QPCR Master Mix (Stratagene) following the manufacturer’s instructions. Amplification was performed using the ABI 7500 Real Time System.
4.10. Western Blot Analyses
Tumor spheres of each sample were collected for extracting protein. Western blot was performed with conventional methods [
16]. Protein samples were separated with 15% SDS-polyacrylamide gel and transferred onto PVDF membranes (Millipore). The primary antibodies used in this study include CD133, EMA, vimentin, VEGF. Blots were washed and incubated for 1 h with IRDyeTM800 conjugated anti-goat and anti-mouse second antibodies.
4.11. Statistical Analysis
Data were analyzed using GraphPad-Prism software (version 5.0; Graphpad software Inc.: San Diego, CA, USA) and Windows XP Excel 2007 (version 2007; Microsoft: Redmond, WA, USA, 2007). Statistical analysis was performed using Student’s t test for mRNA level analysis. Probabilities (p value) < 0.05 were regarded statistically significant.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}