State of the Art in Silico Tools for the Study of Signaling Pathways in Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
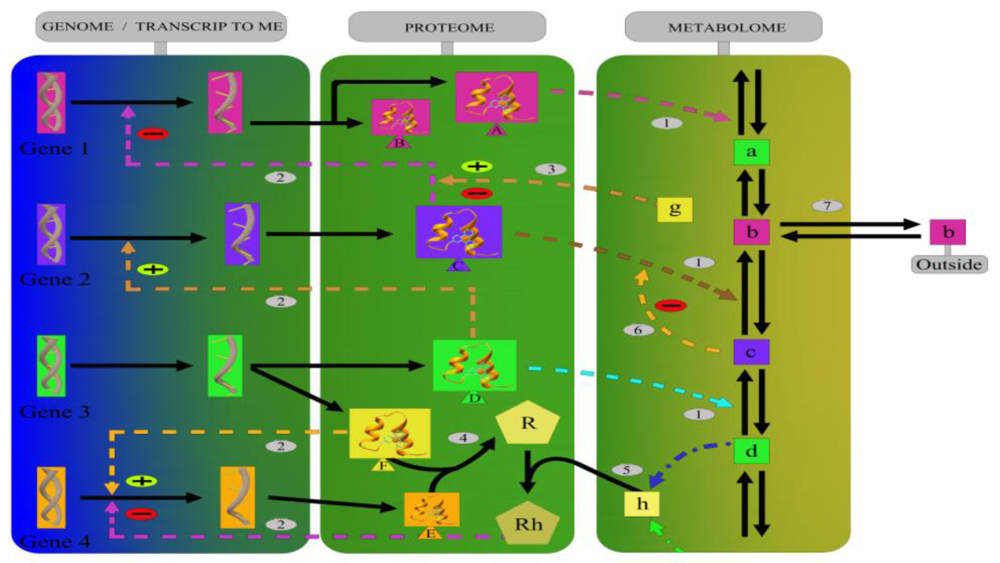
2. The Biochemical Networks in Cancer
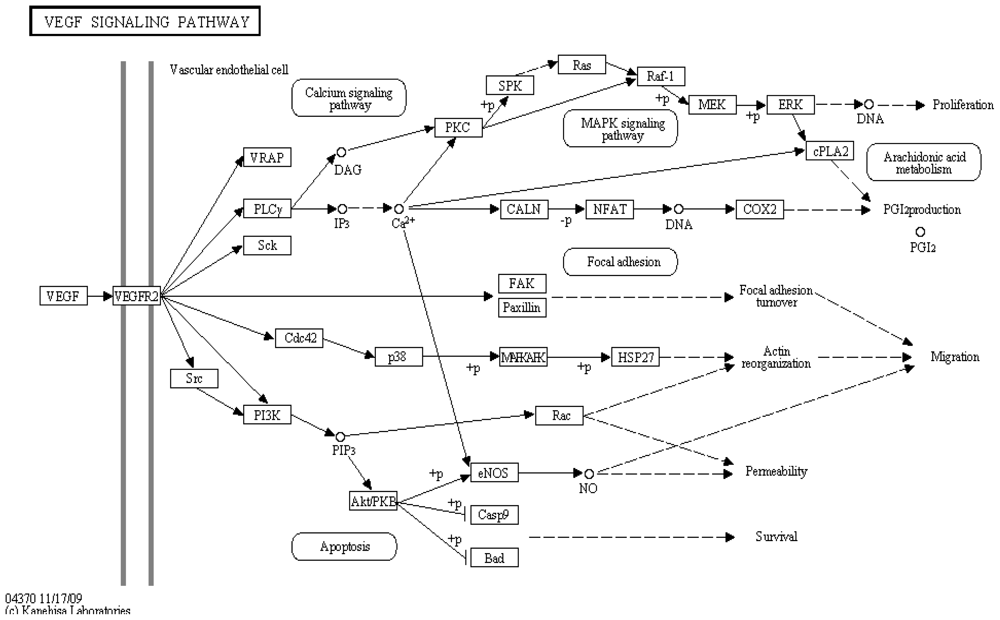
3. Bioinformatic Tools for Pathway Analysis
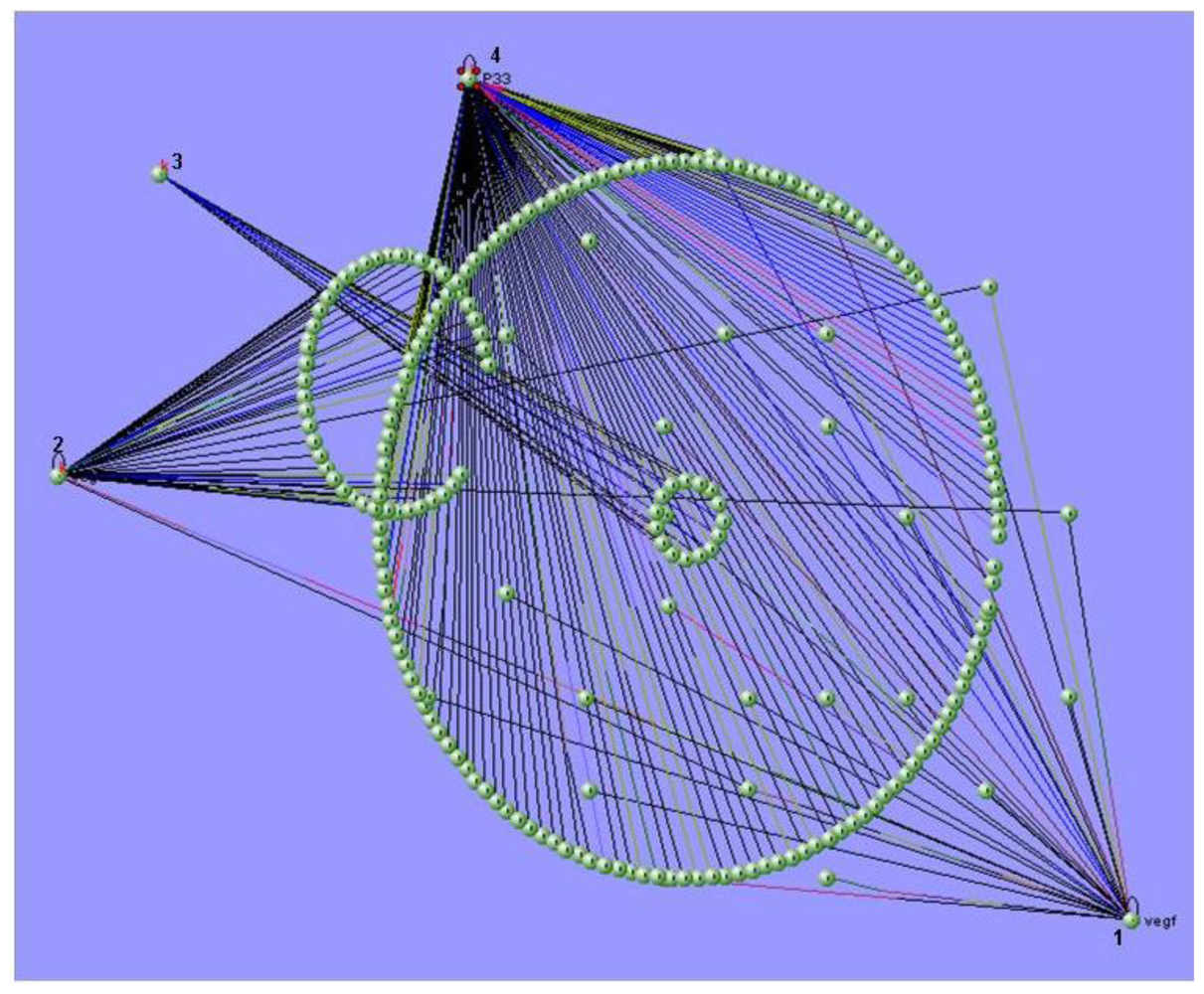
4. Bioinformatics Tools for Systems Biology
5. The Virtual Cell
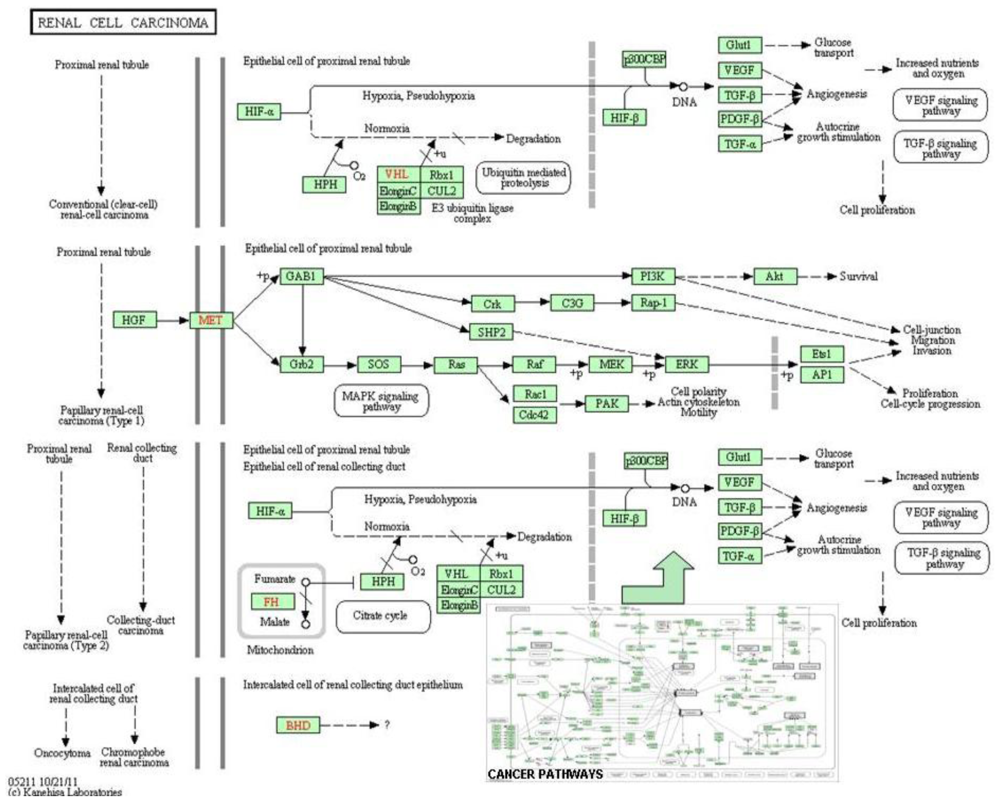
6. Systems Biology and Cancer
7. Conclusions
- Conflict of InterestThe authors have no conflicts of interest to declare.
References
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar]
- Patterson, S.D.; Aebersold, R.H. Proteomics: The first decade and beyond. Nat. Genet 2003, 33, 311–323. [Google Scholar]
- Fiehn, O. Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genomics 2001, 2, 155–168. [Google Scholar]
- Mukherji, S.; van Oudenaarden, A. Synthetic biology: Understanding biological design from synthetic circuits. Nat. Rev. Genet 2009, 10, 859–871. [Google Scholar]
- Vieites, J.M.; Guazzaroni, M.-E.; Beloqui, A.; Golyshin, P.N.; Ferrer, M. Metagenomics approaches in systems microbiology. FEMS Microbiol. Rev 2009, 33, 236–255. [Google Scholar]
- O’Callaghan, P.M.; James, D.C. Systems biotechnology of mammalian cell factories. Brief. Funct. Genomics Proteomics 2008, 7, 95–110. [Google Scholar]
- Tomlins, S.A.; Rubin, M.A.; Chinnaiyan, A.M. Integrative biology of prostate cancer progression. Annu. Rev. Pathol 2006, 1, 243–271. [Google Scholar]
- Liu, E.T. Integrative biology—A strategy for systems biomedicine. Nat. Rev. Genet 2009, 10, 64–68. [Google Scholar]
- Kirschner, M.W. The meaning of systems biology. Cell 2005, 121, 503–504. [Google Scholar]
- Bayer, K.U.; de Koninck, P.; Leonard, A.S.; Hell, J.W.; Schulman, H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 2001, 411, 801–805. [Google Scholar]
- Shimizu, T.S.; le Novere, N.; Levin, M.D.; Beavil, A.J.; Sutton, B.J.; Bray, D. Molecular model of a lattice of signalling proteins involved in bacterial chemotaxis. Nat. Cell. Biol 2000, 2, 792–796. [Google Scholar]
- Arkin, A.; Ross, J.; McAdams, H.H. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics 1998, 149, 1633–1648. [Google Scholar]
- Portin, P. The elusive concept of the gene. Hereditas 2009, 146, 112–117. [Google Scholar]
- Likić, V.A.; McConville, M.J.; Lithgow, T.; Bacic, A. Systems biology: The next frontier for bioinformatics. Adv. Bioinform. [CrossRef]
- Gingeras, T.R. Origin of phenotypes: Genes and transcripts. Genome Res 2007, 17, 682–690. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Gasparini, G.; Longo, R. The paradigm of personalized therapy in oncology. Expert Opin. Ther. Targets 2011, 11. [Google Scholar] [CrossRef]
- Blanco, E.; Hsiao, A.; Ruiz-Esparza, G.U.; Landry, M.G.; Meric-Bernstam, F.; Ferrari, M. Molecular-targeted nanotherapies in cancer: Enabling treatment specificity. Mol. Oncol 2011, 5, 492–503. [Google Scholar]
- Voon, P.J.; Kong, H.L. Tumour genetics and genomics to personalise cancer treatment. Ann. Acad. Med. Singap 2011, 40, 362–367. [Google Scholar]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med 2011, 17, 1359–1370. [Google Scholar]
- Dinicola, S.; D’Anselmi, F.; Pasqualato, A.; Proietti, S.; Lisi, E.; Cucina, A.; Bizzarri, M. A systems biology approach to cancer: Fractals, attractors, and nonlinear dynamics. OMICS 2011, 15, 93–104. [Google Scholar]
- Pérez-Losada, J.; Castellanos-Martín, A.; Mao, J.H. Cancer evolution and individual susceptibility. Integr. Biol. (Camb.) 2011, 3, 316–328. [Google Scholar]
- Tian, T.; Olson, S.; Whitacre, J.M.; Harding, A. The origins of cancer robustness and evolvability. Integr. Biol. (Camb.) 2011, 3, 17–30. [Google Scholar] [Green Version]
- Stambuk, S.; Sundov, D.; Kuret, S.; Beljan, R.; Andelinović, S. Future perspectives of personalized oncology. Coll. Antropol 2010, 34, 763–769. [Google Scholar]
- Hiissa, J.; Elo, L.L.; Huhtinen, K.; Perheentupa, A.; Poutanen, M.; Aittokallio, T. Resampling reveals sample-level differential expression in clinical genome-wide studies. OMICS 2009, 13, 381–396. [Google Scholar]
- Hwang, T.; Park, T. Identification of differentially expressed subnetworks based on multivariate ANOVA. BMC Bioinforma 2009, 10. [Google Scholar] [CrossRef]
- Hornberga, J.J.; Bruggeman, F.J.; Westerhoff, H.V.; Lankelma, J. Cancer: A systems biology disease. BioSystems 2006, 83, 81–90. [Google Scholar]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; Rajewsky, N. Combinatorial miRNA target predictions. Nat. Genet 2005, 37, 495–500. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are miRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar]
- Yu, H.; Luscombe, N.M.; Lu, H.X.; Zhu, X.; Xia, Y.; Han, J.D.; Bertin, N.; Chung, S.; Vidal, M.; Gerstein, M. Annotation transfer between genomes: Protein-protein interologs and protein-DNA regulogs. Genome Res 2004, 14, 1107–1118. [Google Scholar]
- Sharan, R.; Suthram, S.; Kelley, R.M.; Kuhn, T.; McCuine, S.; Uetz, P.; Sittler, T.; Karp, R.M.; Ideker, T. Conserved patterns of protein interaction in multiple species. Proc. Natl. Acad. Sci. USA 2005, 102, 1974–1979. [Google Scholar]
- Shou, C.; Bhardwaj, N.; Lam, H.Y.; Yan, K.K.; Kim, P.M.; Snyder, M.; Gerstein, M.B. Measuring the evolutionary rewiring of biological networks. PLoS Comput. Biol 2011, 7. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Tjian, R. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 1989, 245, 371–378. [Google Scholar]
- Kemp, B.E.; Bylund, D.B.; Huang, T.S.; Krebs, E.G. Substrate specificity of the cyclic AMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1975, 72, 3448–3452. [Google Scholar]
- Lavoie, H.; Hogues, H.; Mallick, J.; Sellam, A.; Nantel, A.; Whiteway, M. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol 2010, 8. [Google Scholar] [CrossRef]
- Amoutzias, G.D.; He, Y.; Gordon, J.; Mossialos, D.; Oliver, S.G.; van de Peer, Y. Posttranslational regulation impacts the fate of duplicated genes. Proc. Natl. Acad. Sci. USA 2010, 107, 2967–2971. [Google Scholar]
- Khatri, P.; Draghici, S. Ontological analysis of gene expression data: Current tools, limitations, and open problems. Bioinformatics 2005, 21, 3587–3595. [Google Scholar]
- Goeman, J.J.; van de Geer, S.A.; de Kort, F.; van Houwelingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar]
- Stelling, J. Mathematical models in microbial systems biology. Curr. Opin. Microbiol 2004, 7, 513–518. [Google Scholar]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1999, 27, 29–34. [Google Scholar]
- Joshi-Tope, G.; Gillespie, M.; Vasrik, I.; D’Eustachio, P.; Schmidt, E.; de Bone, B.; Jassal, B.; Gopinath, G.R.; Wu, G.R.; Matthews, L.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res 2005, 33, D428–D432. [Google Scholar]
- Dahlquist, K.; Salomonis, N.; Vranizan, K.; Lawlor, S.; Conklin, B. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat. Genet 2002, 31, 19–20. [Google Scholar]
- Doniger, S.W.; Salomonis, N.; Dahlquist, K.D.; Vranizan, K.; Lawlor, S.C.; Conklin, B.R. MAPPfinder: Using Gene Ontology and GenMAPP to create a global gene expression profile from microarray data. Genome Biol 2003, 4. [Google Scholar] [CrossRef]
- Grosu, P.; Townsend, J.P.; Hartl, D.L.; Cavalieri, D. Pathway processor: A tool for integrating whole-genome expression results into metabolic networks. Genome Res 2002, 12, 1121–1126. [Google Scholar]
- Pan, D.; Sun, N.; Cheung, K.-H.; Guan, Z.; Ma, L.; Holford, M.; Deng, X.; Zhao, H. PathMAPA: A tool for displaying gene expression and performing statistical tests on metabolic pathways at multiple levels for. Arbidopsis. BMC Bioinforma 2003, 4. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowskis, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar]
- Pandey, R.; Guru, R.K.; Mount, D.W. Pathway miner: Extracting gene association networks from molecular pathways for predicting the biological significance of gene expression microarray data. Bioinformatics 2004, 20, 2156–2158. [Google Scholar]
- Chung, H.-J.; Kim, M.; Park, C.H.; Kim, J.; Kim, J.H. ArrayXPath: Mapping and visualizing microarray gene-expression data with integrated biological pathway resources using scalable vector graphics. Nucleic Acids Res 2004, 32, W460–W464. [Google Scholar]
- Holford, M.; Li, N.; Nadkarni, P.; Zhao, H. VitaPad: Visualization tools for the analysis of pathway data. Bioinformatics 2004, 21, 1596–1602. [Google Scholar]
- Nikitin, A.; Egorov, S.; Daraselia, N.; Mazo, I. Pathway studio-The analysis and navigation of molecular networks. Bioinformatics 2003, 19, 2155–2157. [Google Scholar]
- Krishnappa, R. Molecular expression profiling with respect to KEGG hsa05219 pathway. Ecancermedicalscience 2011, 5. [Google Scholar] [CrossRef]
- Facciotti, M.T.; Bonneau, R.; Hood, L.; Baliga, N.S. Systems biology experimental design—Considerations for building predictive gene regulatory network models for prokaryotic systems. Curr. Genomics 2004, 5, 527–544. [Google Scholar]
- Hu, Z.; Mellor, J.; Wu, J.; DeLisi, C. VisANT: An online visualization and analysis tool for biological interaction data. BMC Bioinforma 2004, 5. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res 2010, 37, D26–D31. [Google Scholar]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res 2006, 34, D302–D305. [Google Scholar]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res 2012, 40, D290–D301. [Google Scholar]
- Caspi, R.; Altman, T.; Dreher, K.; Fulcher, C.A.; Subhraveti, P.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 2012, 40, D742–D53. [Google Scholar]
- Kuhn, M.; Szklarczyk, D.; Franceschini, A.; Campillos, M.; von Mering, C.; Jensen, L.J.; Beyer, A.; Bork, P. STITCH 2: An interaction network database for small molecules and proteins. Nucleic Acids Res 2010, 38, D552–D556. [Google Scholar]
- Lehne, B.; Schlitt, T. Protein-protein interaction databases: Keeping up with growing interactomes. Hum. Genomics 2009, 3, 291–297. [Google Scholar]
- Le Novère, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B.; et al. BioModels database: A free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res 2006, 34, D689–D691. [Google Scholar]
- Karp, P.D.; Paley, S.M.; Krummenacker, M.; Latendresse, M.; Dale, J.M.; Lee, T.J.; Kaipa, P.; Gilham, F.; Spaulding, A.; Popescu, L.; et al. Pathway Tools version 13.0: Integrated software for pathway/genome informatics and systems biology. Brief Bioinform 2010, 11, 40–79. [Google Scholar]
- Merelli, E.; Armano, G.; Cannata, N.; Corradini, F.; d’Inverno, M.; Doms, A.; Lord, P.; Martin, A.; Milanesi, L.; Möller, S.; et al. Agents in bioinformatics, computational and systems biology. Brief Bioinform 2007, 8, 45–59. [Google Scholar]
- Kitano, H. Computational systems biology. Nature 2002, 420, 206–210. [Google Scholar]
- Nurse, P. Universal control mechanism regulating onset of M-phase. Nature 1990, 344, 503–508. [Google Scholar]
- Morgan, D.O. The Cell Cycle: Principles of Control; New Science Press: London, UK, 2006. [Google Scholar]
- Csikász-Nagy, A. Computational systems biology of the cell cycle. Brief. Bioinform 2009, 10, 424–434. [Google Scholar]
- Chi, Y.; Welcker, M.; Hizli, A.A.; Posakony, J.J.; Aebersold, R.; Clurman, B.E. Identification of CDK2 substrates in human cell lysates. Genome Biol 2008, 9. [Google Scholar] [CrossRef]
- Dematte, L.; Priami, C.; Romanel, A. The beta workbench: A computational tool to study the dynamics of biological systems. Brief. Bioinform 2008, 9, 437–449. [Google Scholar]
- Vass, M.; Allen, N.; Shaffer, C.A.; Ramakrishnan, N.; Watson, L.T.; Tyson, J.J. The JigCell model builder and run manager. Bioinformatics 2004, 20, 3680–3681. [Google Scholar]
- Lecca, P.; Palmisano, A.; Priami, C. A New Probabilistic Generative Model of Parameter Inference in Biochemical Networks. Proceedings of the 2009 ACM Symposium on Applied Computing, Honolulu, HI, USA, 8–12 March 2009.
- Zi, Z.; Klipp, E. SBML-PET: A systems biology markup language-based parameter estimation tool. Bioinformatics 2006, 22, 2704–2705. [Google Scholar]
- Zwolak, J.W.; Tyson, J.J.; Watson, L.T. Globally optimized parameters for a model of mitotic control in frog egg extracts. Syst. Biol 2005, 152, 81–92. [Google Scholar]
- Baumbach, J.; Apeltsin, L. Linking Cytoscape and the corynebacterial reference database CoryneRegNet. BMC Genomics 2008, 9. [Google Scholar] [CrossRef]
- Andrecut, M.; Huang, S.; Kauffman, S.A. Heuristic approach to sparse approximation of gene regulatory networks. J. Comput. Biol 2008, 15, 1173–1186. [Google Scholar]
- Cheng, C.; Li, L.M. Systematic identification of cell cycle regulated transcription factors from microarray time series data. BMC Genomics 2008, 9. [Google Scholar] [CrossRef]
- Wu, W.S.; Li, W.H. Systematic identification of yeast cell cycle transcription factors using multiple data sources. BMC Bioinforma 2008, 9. [Google Scholar] [CrossRef]
- Yang, L.; Han, Z.; Robb MacLellan, W.; Weiss, J.N.; Qu, Z. Linking cell division to cell growth in a spatiotemporal model of the cell cycle. J. Theor. Biol 2006, 241, 120–133. [Google Scholar]
- Conradi, C.; Flockerzi, D.; Raisch, J.; Stelling, J. Subnetwork analysis reveals dynamic features of complex (bio)chemical networks. Proc. Natl. Acad. Sci. USA 2007, 104, 19175–19180. [Google Scholar]
- Lovrics, A.; Csikász-Nagy, A.; Zsély, I.G.; Zádor, J.; Turányi, T.; Novák, B. Time scale and dimension analysis of a budding yeast cell cycle model. BMC Bioinforma 2006, 7. [Google Scholar] [CrossRef]
- Anderson, A.R.; Quaranta, V. Integrative mathematical oncology. Nat. Rev. Cancer 2008, 8, 227–234. [Google Scholar]
- Araujo, R.P.; McElwain, D.L. A history of the study of solid tumour growth: The contribution of mathematical modelling. Bull. Math. Biol 2004, 66, 1039–1091. [Google Scholar]
- Preziosi, L. Cancer Modelling and Simulation; CRC Press: London, UK, 2003. [Google Scholar]
- Byrne, H.; Drasdo, D. Individual-based and continuum models of growing cell populations: A comparison. J. Math. Biol 2009, 58, 657–687. [Google Scholar]
- Sherratt, J.A.; Chaplain, M.A. A new mathematical model for avascular tumour growth. J. Math. Biol 2001, 43, 291–312. [Google Scholar]
- Schaff, J.; Fink, C.C.; Slepchenko, B.; Carson, J.H.; Loew, L.M. A general computational framework for modeling cellular structure and function. Biophys. J 1997, 73, 1135–1146. [Google Scholar]
- Laubenbacher, R.; Hower, V.; Jarrah, A.; Torti, S.V.; Shulaev, V.; Mendes, P.; Torti, F.M.; Akman, S. A systems biology view of cancer. Biochim. Biophys. Acta 2009, 1796, 129–139. [Google Scholar]
- Daskalaki, A.; Wierling, C.; Herwig, R. Computational Tools and Resources for Systems Biology Approaches to Cancer. In Computational Biology: Issues and Applications in Oncology; Pham, T., Ed.; Springer: New York, NY, USA, 2009; pp. 227–242. [Google Scholar]
- Anderson, A.R.A.; Quaranta, V. Integrative mathematical oncology. Nat. Rev. Cancer 2008, 8, 227–234. [Google Scholar]
- Bauer-Mehren, A.; Rautschka, M.; Sanz, F.; Furlong, L.I. DisGeNET: A Cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics 2010, 26, 2924–2926. [Google Scholar]







© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Villaamil, V.M.; Gallego, G.A.; Cainzos, I.S.; Valladares-Ayerbes, M.; Aparicio, L.M.A. State of the Art in Silico Tools for the Study of Signaling Pathways in Cancer. Int. J. Mol. Sci. 2012, 13, 6561-6581. https://doi.org/10.3390/ijms13066561
Villaamil VM, Gallego GA, Cainzos IS, Valladares-Ayerbes M, Aparicio LMA. State of the Art in Silico Tools for the Study of Signaling Pathways in Cancer. International Journal of Molecular Sciences. 2012; 13(6):6561-6581. https://doi.org/10.3390/ijms13066561
Chicago/Turabian StyleVillaamil, Vanessa Medina, Guadalupe Aparicio Gallego, Isabel Santamarina Cainzos, Manuel Valladares-Ayerbes, and Luis M. Antón Aparicio. 2012. "State of the Art in Silico Tools for the Study of Signaling Pathways in Cancer" International Journal of Molecular Sciences 13, no. 6: 6561-6581. https://doi.org/10.3390/ijms13066561
APA StyleVillaamil, V. M., Gallego, G. A., Cainzos, I. S., Valladares-Ayerbes, M., & Aparicio, L. M. A. (2012). State of the Art in Silico Tools for the Study of Signaling Pathways in Cancer. International Journal of Molecular Sciences, 13(6), 6561-6581. https://doi.org/10.3390/ijms13066561