Neuroglobin, a Novel Target for Endogenous Neuroprotection against Stroke and Neurodegenerative Disorders

Abstract
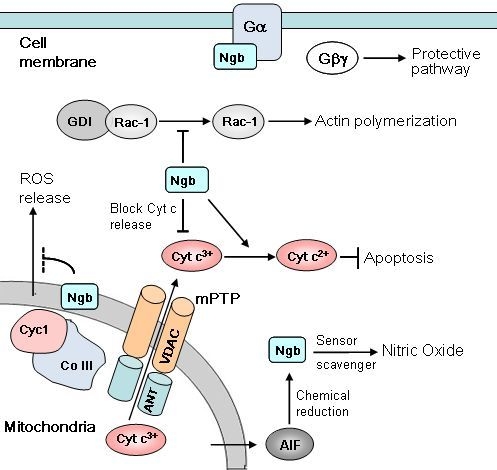
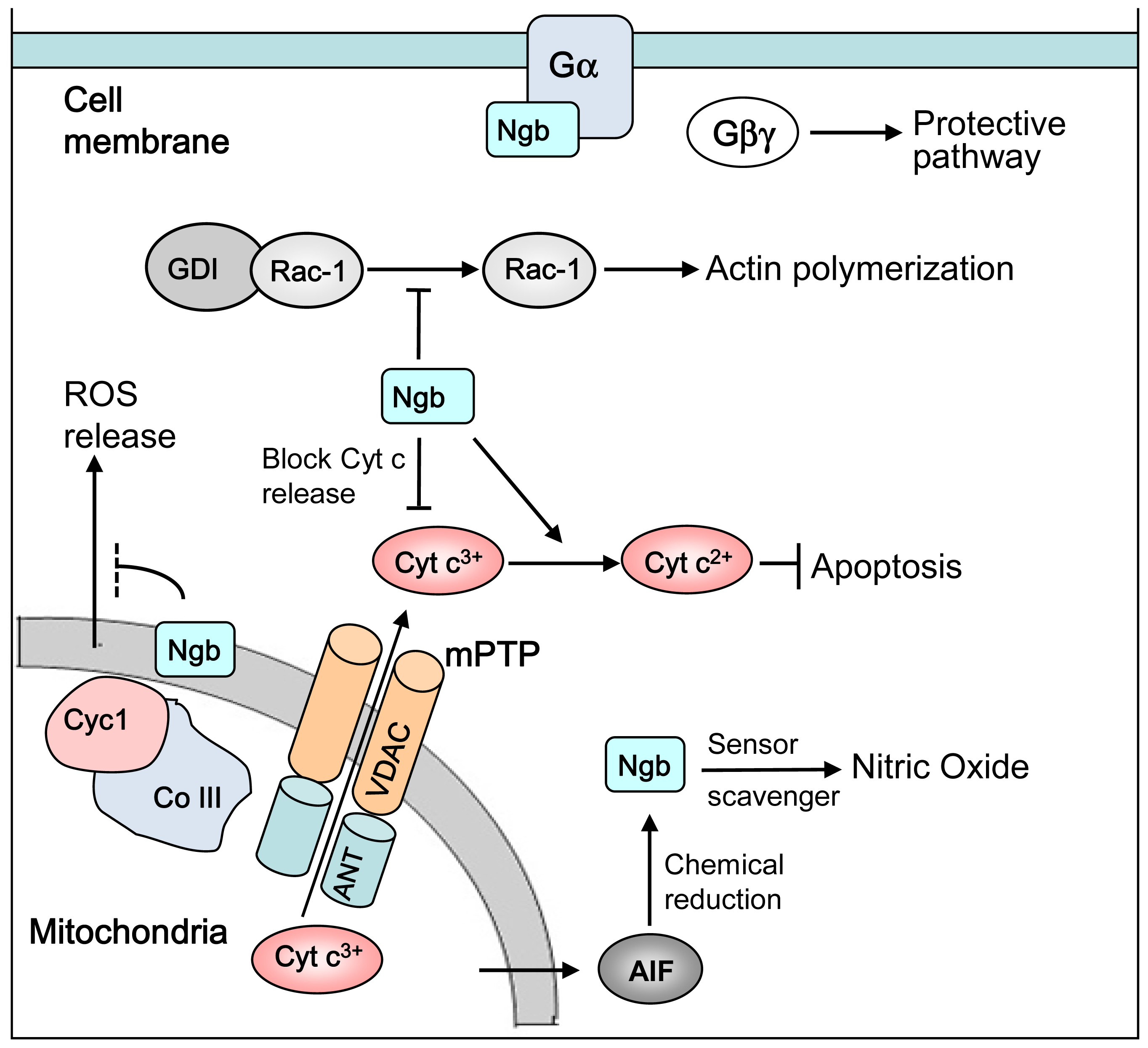
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ngb Can Serve as a Target for Development of Therapeutics against Stroke and Neurodegenerative Disorders
3. Neuroprotective Roles of Ngb against Hypoxic/Ischemic and Oxidative Stress-Related Brain/Neuron Injury
4. Neuroprotective Effects of Ngb against Neurodegenerative and Other Neurological Disorders
5. Molecular Mechanisms of Ngb Neuroprotection
5.1. Oxygen Sensing and ROS Scavenging by Ngb
5.2. Regulation of Signal Transduction
5.3. Maintenance of Mitochondrial Function
6. Tissue Specific Expression of Ngb Gene and Its Regulation
6.1. Neuron-Specific Expression of Ngb Gene
6.2. Ngb Gene Expression under Pathological Conditions
6.3. Transcriptional Regulation of Ngb Gene
6.4. Ngb Gene Expression in Non-Neuronal Cells
7. Targeting Ngb for Endogenous Neuroprotection
7.1. Previously Identified Chemical Compounds that Up-Regulate Endogenous Ngb Expression
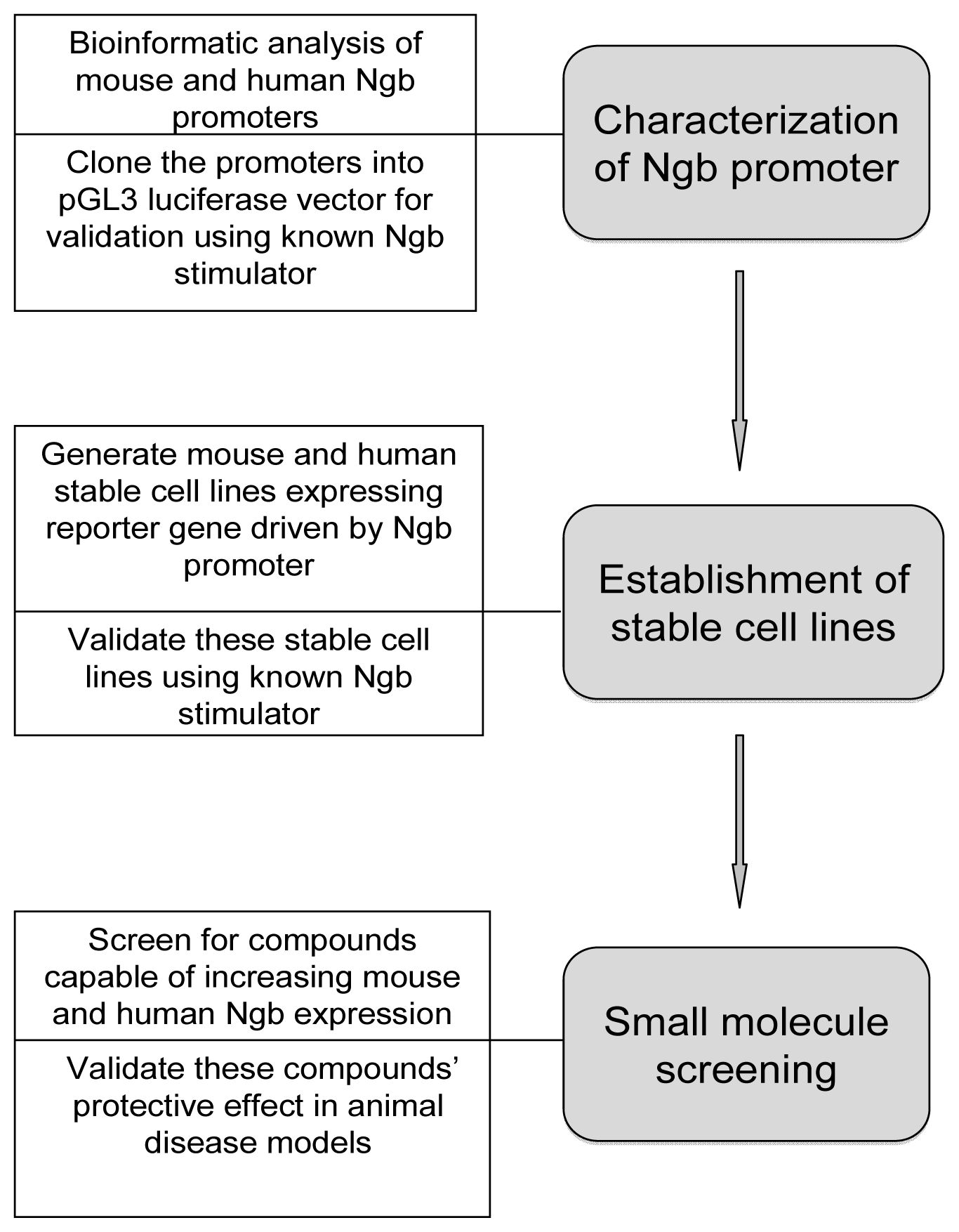
7.2. Establishment of a Cell-based High Throughput Screening System for Identification of Small Molecules Capable of Ngb Upregulation
Acknowledgments
References
- Burmester, T.; Weich, B.; Reinhardt, S.; Hankeln, T. A vertebrate globin expressed in the brain. Nature 2000, 407, 520–523. [Google Scholar]
- Sun, Y.; Jin, K.; Mao, X.O.; Zhu, Y.; Greenberg, D.A. Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc. Natl. Acad. Sci. USA 2001, 98, 15306–15311. [Google Scholar]
- Peroni, D.; Negro, A.; Bahr, M.; Dietz, G.P. Intracellular delivery of neuroglobin using hiv-1 tat protein transduction domain fails to protect against oxygen and glucose deprivation. Neurosci. lett 2007, 421, 110–114. [Google Scholar]
- Hundahl, C.; Kelsen, J.; Kjaer, K.; Ronn, L.C.; Weber, R.E.; Geuens, E.; Hay-Schmidt, A.; Nyengaard, J.R. Does neuroglobin protect neurons from ischemic insult? A quantitative investigation of neuroglobin expression following transient mcao in spontaneously hypertensive rats. Brain Res 2006, 1085, 19–27. [Google Scholar]
- Wang, X.; Liu, J.; Zhu, H.; Tejima, E.; Tsuji, K.; Murata, Y.; Atochin, D.N.; Huang, P.L.; Zhang, C.; Lo, E.H. Effects of neuroglobin overexpression on acute brain injury and long-term outcomes after focal cerebral ischemia. Stroke 2008, 39, 1869–1874. [Google Scholar]
- Sun, Y.; Jin, K.; Peel, A.; Mao, X.O.; Xie, L.; Greenberg, D.A. Neuroglobin protects the brain from experimental stroke in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 3497–3500. [Google Scholar]
- Khan, A.A.; Mao, X.O.; Banwait, S.; Jin, K.; Greenberg, D.A. Neuroglobin attenuates beta-amyloid neurotoxicity in vitro and transgenic alzheimer phenotype in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19114–19119. [Google Scholar]
- Mergenthaler, P.; Dirnagl, U. Protective conditioning of the brain: Expressway or roadblock? J. Physiol 2011, 589, 4147–4155. [Google Scholar]
- Manuvakhova, M.S.; Johnson, G.G.; White, M.C.; Ananthan, S.; Sosa, M.; Maddox, C.; McKellip, S.; Rasmussen, L.; Wennerberg, K.; Hobrath, J.V.; et al. Identification of novel small molecule activators of nuclear factor-κb with neuroprotective action via high-throughput screening. J. Neurosci. Res 2011, 89, 58–72. [Google Scholar]
- Sun, Y.; Jin, K.; Mao, X.O.; Zhu, Y.; Greenberg, D.A. Neuroglobin protects the brain from experimental stroke in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 15306–15311. [Google Scholar]
- Garry, D.J.; Mammen, P.P. Neuroprotection and the role of neuroglobin. Lancet 2003, 362, 342–343. [Google Scholar]
- Greenberg, D.A.; Jin, K.; Khan, A.A. Neuroglobin: An endogenous neuroprotectant. Curr. Opin. Pharmacol 2008, 8, 20–24. [Google Scholar]
- Yu, Z.; Fan, X.; Lo, E.H.; Wang, X. Neuroprotective roles and mechanisms of neuroglobin. Neurol. Res 2009, 31, 122–127. [Google Scholar]
- Venis, S. Neuroglobin might protect brain cells during stroke. Lancet 2001, 358(01), 07148–3. [Google Scholar] [CrossRef]
- Watanabe, S.; Wakasugi, K. Zebrafish neuroglobin is a cell-membrane-penetrating globin. Biochemistry 2008, 47, 5266–5270. [Google Scholar]
- Jin, K.; Mao, X.O.; Xie, L.; John, V.; Greenberg, D.A. Pharmacological induction of neuroglobin expression. Pharmacology 2011, 87, 81–84. [Google Scholar]
- de Marinis, E.; Ascenzi, P.; Pellegrini, M.; Galluzzo, P.; Bulzomi, P.; Arevalo, M.A.; Garcia-Segura, L.M.; Marino, M. 17β-estradiol—A new modulator of neuroglobin levels in neurons: Role in neuroprotection against H2O2-induced toxicity. Neurosignals 2010, 18, 223–235. [Google Scholar]
- Reuss, S.; Saaler-Reinhardt, S.; Weich, B.; Wystub, S.; Reuss, M.H.; Burmester, T.; Hankeln, T. Expression analysis of neuroglobin mrna in rodent tissues. Neuroscience 2002, 115, 645–656. [Google Scholar]
- Nienhaus, K.; Nienhaus, G.U. Searching for neuroglobin’s role in the brain. IUBMB Life 2007, 59, 490–497. [Google Scholar]
- Brunori, M.; Vallone, B. A globin for the brain. FASEB J 2006, 20, 2192–2197. [Google Scholar]
- Burmester, T.; Hankeln, T. Neuroglobin: A respiratory protein of the nervous system. News Physiol. Sci 2004, 19, 110–113. [Google Scholar]
- Greenberg, D.A.; Jin, K.; Khan, A.A. Neuroglobin: An endogenous neuroprotectant. Curr. Opin. Pharmacol 2008, 8, 20–24. [Google Scholar]
- Giuffre, A.; Moschetti, T.; Vallone, B.; Brunori, M. Is neuroglobin a signal transducer? IUBMB Life 2008, 60, 410–413. [Google Scholar]
- Brunori, M.; Vallone, B. Neuroglobin, seven years after. Cell Mol. Life Sci 2007, 64, 1259–1268. [Google Scholar]
- Fordel, E.; Thijs, L.; Martinet, W.; Schrijvers, D.; Moens, L.; Dewilde, S. Anoxia or oxygen and glucose deprivation in sh-sy5y cells: A step closer to the unraveling of neuroglobin and cytoglobin functions. Gene 2007, 398, 114–122. [Google Scholar]
- Khan, A.A.; Wang, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Miles, E.; Graboski, J.; Chen, S.; Ellerby, L.M.; Jin, K.; et al. Neuroglobin-overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc. Natl. Acad. Sci. USA 2006, 103, 17944–17948. [Google Scholar]
- Cai, B.; Lin, Y.; Xue, X.H.; Fang, L.; Wang, N.; Wu, Z.Y. Tat-mediated delivery of neuroglobin protects against focal cerebral ischemia in mice. Exp. Neurol 2011, 227, 224–231. [Google Scholar]
- Li, R.C.; Pouranfar, F.; Lee, S.K.; Morris, M.W.; Wang, Y.; Gozal, D. Neuroglobin protects pc12 cells against β-amyloid-induced cell injury. Neurobiol. Aging 2008, 29, 1815–1822. [Google Scholar]
- Lima, D.C.; Cossa, A.C.; Perosa, S.R.; de Oliveira, E.M.; da Silva, J.A.J.; da Silva Fernandes, M.J.; da Silva, I.R.; Higa, E.M.; da Graca Naffah-Mazzacoratti, M.; Cavalheiro, E.A.; et al. Neuroglobin is up-regulated in the cerebellum of pups exposed to maternal epileptic seizures. Int. J. Dev. Neurosci 2011, 29, 891–897. [Google Scholar]
- Wei, X.; Yu, Z.; Cho, K.S.; Chen, H.; Malik, M.T.; Chen, X.; Lo, E.H.; Wang, X.; Chen, D.F. Neuroglobin is an endogenous neuroprotectant for retinal ganglion cells against glaucomatous damage. Am. J. Pathol 2011, 179, 2788–2797. [Google Scholar]
- Pesce, A.; Dewilde, S.; Nardini, M.; Moens, L.; Ascenzi, P.; Hankeln, T.; Burmester, T.; Bolognesi, M. Human brain neuroglobin structure reveals a distinct mode of controlling oxygen affinity. Structure 2003, 11, 1087–1095. [Google Scholar]
- Vallone, B.; Nienhaus, K.; Brunori, M.; Nienhaus, G.U. The structure of murine neuroglobin: Novel pathways for ligand migration and binding. Proteins 2004, 56, 85–92. [Google Scholar]
- Wystub, S.; Laufs, T.; Schmidt, M.; Burmester, T.; Maas, U.; Saaler-Reinhardt, S.; Hankeln, T.; Reuss, S. Localization of neuroglobin protein in the mouse brain. Neurosci. Lett 2003, 346, 114–116. [Google Scholar]
- Pesce, A.; Dewilde, S.; Nardini, M.; Moens, L.; Ascenzi, P.; Hankeln, T.; Burmester, T.; Bolognesi, M. The human brain hexacoordinated neuroglobin three-dimensional structure. Micron 2004, 35, 63–65. [Google Scholar]
- Kriegl, J.M.; Bhattacharyya, A.J.; Nienhaus, K.; Deng, P.; Minkow, O.; Nienhaus, G.U. Ligand binding and protein dynamics in neuroglobin. Proc. Natl. Acad. Sci. USA 2002, 99, 7992–7997. [Google Scholar]
- Fago, A.; Hundahl, C.; Dewilde, S.; Gilany, K.; Moens, L.; Weber, R.E. Allosteric regulation and temperature dependence of oxygen binding in human neuroglobin and cytoglobin. Molecular mechanisms and physiological significance. J. Biol. Chem 2004, 279, 44417–44426. [Google Scholar]
- Van Doorslaer, S.; Dewilde, S.; Kiger, L.; Nistor, S.V.; Goovaerts, E.; Marden, M.C.; Moens, L. Nitric oxide binding properties of neuroglobin. A characterization by epr and flash photolysis. J. Biol. Chem 2003, 278, 4919–4925. [Google Scholar]
- Hundahl, C.A.; Kelsen, J.; Dewilde, S.; Hay-Schmidt, A. Neuroglobin in the rat brain (ii): Co-localisation with neurotransmitters. Neuroendocrinology 2008, 88, 183–198. [Google Scholar]
- Brunori, M.; Giuffre, A.; Nienhaus, K.; Nienhaus, G.U.; Scandurra, F.M.; Vallone, B. Neuroglobin, nitric oxide, and oxygen: Functional pathways and conformational changes. Proc. Natl. Acad. Sci. USA 2005, 102, 8483–8488. [Google Scholar]
- Moncada, S.; Erusalimsky, J.D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol 2002, 3, 214–220. [Google Scholar]
- Brunori, M.; Giuffre, A.; Forte, E.; Mastronicola, D.; Barone, M.C.; Sarti, P. Control of cytochrome c oxidase activity by nitric oxide. Biochim. Biophys. Acta 2004, 1655, 365–371. [Google Scholar]
- Jin, K.; Mao, X.O.; Xie, L.; Khan, A.A.; Greenberg, D.A. Neuroglobin protects against nitric oxide toxicity. Neurosci. Lett 2008, 430, 135–137. [Google Scholar]
- Fordel, E.; Thijs, L.; Martinet, W.; Lenjou, M.; Laufs, T.; van Bockstaele, D.; Moens, L.; Dewilde, S. Neuroglobin and cytoglobin overexpression protects human sh-sy5y neuroblastoma cells against oxidative stress-induced cell death. Neurosci. Lett 2006, 410, 146–151. [Google Scholar]
- Sun, Y.; Jin, K.; Mao, X.O.; Xie, L.; Peel, A.; Childs, J.T.; Logvinova, A.; Wang, X.; Greenberg, D.A. Effect of aging on neuroglobin expression in rodent brain. Neurobiol. Aging 2005, 26, 275–278. [Google Scholar]
- Wakasugi, K.; Nakano, T.; Morishima, I. Oxidized human neuroglobin acts as a heterotrimeric galpha protein guanine nucleotide dissociation inhibitor. J. Biol. Chem 2003, 278, 36505–36512. [Google Scholar]
- Schwindinger, W.F.; Robishaw, J.D. Heterotrimeric g-protein βγ-dimers in growth and differentiation. Oncogene 2001, 20, 1653–1660. [Google Scholar]
- Watanabe, S.; Wakasugi, K. Neuroprotective function of human neuroglobin is correlated with its guanine nucleotide dissociation inhibitor activity. Biochem. Biophys. Res. Commun 2008, 369, 695–700. [Google Scholar]
- Khan, A.A.; Mao, X.O.; Banwait, S.; DerMardirossian, C.M.; Bokoch, G.M.; Jin, K.; Greenberg, D.A. Regulation of hypoxic neuronal death signaling by neuroglobin. FASEB J 2008, 22, 1737–1747. [Google Scholar]
- Wakasugi, K.; Nakano, T.; Kitatsuji, C.; Morishima, I. Human neuroglobin interacts with flotillin-1, a lipid raft microdomain-associated protein. Biochem. Biophys. Res. Commun 2004, 318, 453–460. [Google Scholar]
- Wakasugi, K.; Nakano, T.; Morishima, I. Association of human neuroglobin with cystatin c, a cysteine proteinase inhibitor. Biochemistry 2004, 43, 5119–5125. [Google Scholar]
- Tiso, M.; Tejero, J.; Basu, S.; Azarov, I.; Wang, X.; Simplaceanu, V.; Frizzell, S.; Jayaraman, T.; Geary, L.; Shapiro, C.; et al. Human neuroglobin functions as a redox-regulated nitrite reductase. J. Biol. Chem 2011, 286, 18277–18289. [Google Scholar]
- Giuffre, A.; Moschetti, T.; Vallone, B.; Brunori, M. Neuroglobin: Enzymatic reduction and oxygen affinity. Biochem. Biophys. Res. Commun 2008, 367, 893–898. [Google Scholar]
- Modjtahedi, N.; Giordanetto, F.; Madeo, F.; Kroemer, G. Apoptosis-inducing factor: Vital and lethal. Trends Cell Biol 2006, 16, 264–272. [Google Scholar]
- Fago, A.; Mathews, A.J.; Moens, L.; Dewilde, S.; Brittain, T. The reaction of neuroglobin with potential redox protein partners cytochrome b5 and cytochrome c. FEBS Lett 2006, 580, 4884–4888. [Google Scholar]
- Yu, Z.; Liu, J.; Guo, S.; Xing, C.; Fan, X.; Ning, M.; Yuan, J.C.; Lo, E.H.; Wang, X. Neuroglobin-overexpression alters hypoxic response gene expression in primary neuron culture following oxygen glucose deprivation. Neuroscience 2009, 162, 396–403. [Google Scholar]
- Chen, L.M.; Xiong, Y.S.; Kong, F.L.; Qu, M.; Wang, Q.; Chen, X.Q.; Wang, J.Z.; Zhu, L.Q. Neuroglobin attenuates alzheimer-like tau hyperphosphorylation by activating akt signaling. J. Neurochem 2012, 120, 157–164. [Google Scholar]
- Burmester, T.; Gerlach, F.; Hankeln, T. Regulation and role of neuroglobin and cytoglobin under hypoxia. Adv. Exp. Med. Biol 2007, 618, 169–180. [Google Scholar]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev 2000, 80, 315–360. [Google Scholar]
- Sims, N.R.; Anderson, M.F. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int 2002, 40, 511–526. [Google Scholar]
- Li, R.C.; Pouranfar, F.; Lee, S.K.; Morris, M.W.; Wang, Y.; Gozal, D. Neuroglobin protects pc12 cells against beta-amyloid-induced cell injury. Neurobiol. Aging 2008, 29, 1815–1822. [Google Scholar]
- Liu, J.; Yu, Z.; Guo, S.; Lee, S.R.; Xing, C.; Zhang, C.; Gao, Y.; Nicholls, D.G.; Lo, E.H.; Wang, X. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. J. Neurosci. Res 2009, 87, 164–170. [Google Scholar]
- Saito, A.; Maier, C.M.; Narasimhan, P.; Nishi, T.; Song, Y.S.; Yu, F.; Liu, J.; Lee, Y.S.; Nito, C.; Kamada, H.; et al. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol. Neurobiol 2005, 31, 105–116. [Google Scholar]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metabol 2001, 21, 2–14. [Google Scholar]
- Perez-Pinzon, M.A.; Dave, K.R.; Raval, A.P. Role of reactive oxygen species and protein kinase c in ischemic tolerance in the brain. Antioxid. Redox. Signal 2005, 7, 1150–1157. [Google Scholar]
- Larsen, G.A.; Skjellegrind, H.K.; Berg-Johnsen, J.; Moe, M.C.; Vinje, M.L. Depolarization of mitochondria in isolated ca1 neurons during hypoxia, glucose deprivation and glutamate excitotoxicity. Brain Res 2006, 1077, 153–160. [Google Scholar]
- Zhang, W.H.; Wang, H.; Wang, X.; Narayanan, M.V.; Stavrovskaya, I.G.; Kristal, B.S.; Friedlander, R.M. Nortriptyline protects mitochondria and reduces cerebral ischemia/hypoxia injury. Stroke 2008, 39, 455–462. [Google Scholar]
- Zhu, S.; Stavrovskaya, I.G.; Drozda, M.; Kim, B.Y.; Ona, V.; Li, M.; Sarang, S.; Liu, A.S.; Hartley, D.M.; Wu, D.C.; et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002, 417, 74–78. [Google Scholar]
- Susin, S.A.; Zamzami, N.; Castedo, M.; Hirsch, T.; Marchetti, P.; Macho, A.; Daugas, E.; Geuskens, M.; Kroemer, G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med 1996, 184, 1331–1341. [Google Scholar]
- Petit, P.X.; Goubern, M.; Diolez, P.; Susin, S.A.; Zamzami, N.; Kroemer, G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: The impact of irreversible permeability transition. FEBS Lett 1998, 426, 111–116. [Google Scholar]
- Yu, Z.; Liu, N.; Wang, Y.; Li, X.; Wang, X. Identification of neuroglobin-interacting proteins using yeast two-hybrid screening. Neuroscience 2012, 200, 99–105. [Google Scholar]
- Sun, J.; Trumpower, B.L. Superoxide anion generation by the cytochrome bc1 complex. Arch. Biochem. Biophys 2003, 419, 198–206. [Google Scholar]
- Berry, E.A.; Guergova-Kuras, M.; Huang, L.S.; Crofts, A.R. Structure and function of cytochrome bc complexes. Annu. Rev. Biochem 2000, 69, 1005–1075. [Google Scholar]
- Hunte, C.; Palsdottir, H.; Trumpower, B.L. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett 2003, 545, 39–46. [Google Scholar]
- Klimova, T.; Chandel, N.S. Mitochondrial complex iii regulates hypoxic activation of hif. Cell Death Differ 2008, 15, 660–666. [Google Scholar]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ros production and cellular oxygen sensing. Cell Metabol 2005, 1, 401–408. [Google Scholar]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ros but not oxidative phosphorylation. Cell Metabol 2005, 1, 409–414. [Google Scholar]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 1998, 281, 64–71. [Google Scholar]
- Madesh, M.; Hajnoczky, G. Vdac-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol 2001, 155, 1003–1015. [Google Scholar]
- Billen, L.P.; Kokoski, C.L.; Lovell, J.F.; Leber, B.; Andrews, D.W. Bcl-xl inhibits membrane permeabilization by competing with bax. PLoS Biol 2008, 6. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, C.; Deng, M.; Li, L.; Wang, H.; Fan, M.; Xu, W.; Meng, F.; Qian, L.; He, F. Full-length cdna cloning of human neuroglobin and tissue expression of rat neuroglobin. Biochem. Biophys. Res. Commun 2002, 290, 1411–1419. [Google Scholar]
- Geuens, E.; Brouns, I.; Flamez, D.; Dewilde, S.; Timmermans, J.P.; Moens, L. A globin in the nucleus! J. Biol. Chem 2003, 278, 30417–30420. [Google Scholar]
- Schmidt, M.; Giessl, A.; Laufs, T.; Hankeln, T.; Wolfrum, U.; Burmester, T. How does the eye breathe? Evidence for neuroglobin-mediated oxygen supply in the mammalian retina. J. Biol. Chem 2003, 278, 1932–1935. [Google Scholar]
- Hundahl, C.; Stoltenberg, M.; Fago, A.; Weber, R.E.; Dewilde, S.; Fordel, E.; Danscher, G. Effects of short-term hypoxia on neuroglobin levels and localization in mouse brain tissues. Neuropathol. Appl. Neurobiol 2005, 31, 610–617. [Google Scholar]
- Schmidt-Kastner, R.; Haberkamp, M.; Schmitz, C.; Hankeln, T.; Burmester, T. Neuroglobin mrna expression after transient global brain ischemia and prolonged hypoxia in cell culture. Brain Res 2006, 1103, 173–180. [Google Scholar]
- Shao, G.; Gong, K.R.; Li, J.; Xu, X.J.; Gao, C.Y.; Zeng, X.Z.; Lu, G.W.; Huo, X. Antihypoxic effects of neuroglobin in hypoxia-preconditioned mice and sh-sy5y cells. Neurosignals 2009, 17, 196–202. [Google Scholar]
- Shang, A.; Zhou, D.; Wang, L.; Gao, Y.; Fan, M.; Wang, X.; Zhou, R.; Zhang, C. Increased neuroglobin levels in the cerebral cortex and serum after ischemia-reperfusion insults. Brain Res 2006, 1078, 219–226. [Google Scholar]
- Fordel, E.; Thijs, L.; Moens, L.; Dewilde, S. Neuroglobin and cytoglobin expression in mice. Evidence for a correlation with reactive oxygen species scavenging. FEBS J 2007, 274, 1312–1317. [Google Scholar]
- Jin, K.; Mao, Y.; Mao, X.; Xie, L.; Greenberg, D.A. Neuroglobin expression in ischemic stroke. Stroke 2010, 41, 557–559. [Google Scholar]
- Mammen, P.P.; Shelton, J.M.; Goetsch, S.C.; Williams, S.C.; Richardson, J.A.; Garry, M.G.; Garry, D.J. Neuroglobin, a novel member of the globin family, is expressed in focal regions of the brain. J. Histochem. Cytochem 2002, 50, 1591–1598. [Google Scholar]
- Li, R.C.; Lee, S.K.; Pouranfar, F.; Brittian, K.R.; Clair, H.B.; Row, B.W.; Wang, Y.; Gozal, D. Hypoxia differentially regulates the expression of neuroglobin and cytoglobin in rat brain. Brain Res 2006, 1096, 173–179. [Google Scholar]
- Zhang, W.; Tian, Z.; Sha, S.; Cheng, L.Y.; Philipsen, S.; Tan-Un, K.C. Functional and sequence analysis of human neuroglobin gene promoter region. Biochim. Biophys. Acta 2011, 1809, 236–244. [Google Scholar]
- Liu, N.; Yu, Z.; Xiang, S.; Zhao, S.; Tjarnlund-Wolf, A.; Xing, C.; Zhang, J.; Wang, X. Transcriptional regulation mechanisms of hypoxia-induced neuroglobin gene expression. Biochem. J 2012, 443, 153–164. [Google Scholar]
- Hankeln, T.; Wystub, S.; Laufs, T.; Schmidt, M.; Gerlach, F.; Saaler-Reinhardt, S.; Reuss, S.; Burmester, T. The cellular and subcellular localization of neuroglobin and cytoglobin—A clue to their function? IUBMB Life 2004, 56, 671–679. [Google Scholar]
- Chen, X.Q.; Qin, L.Y.; Zhang, C.G.; Yang, L.T.; Gao, Z.; Liu, S.; Lau, L.T.; Fung, Y.W.; Greenberg, D.A.; Yu, A.C. Presence of neuroglobin in cultured astrocytes. Glia 2005, 50, 182–186. [Google Scholar]
- Emara, M.; Turner, A.R.; Allalunis-Turner, J. Hypoxic regulation of cytoglobin and neuroglobin expression in human normal and tumor tissues. Cancer Cell Int 2010, 10. [Google Scholar] [CrossRef]
- Emara, M.; Salloum, N.; Allalunis-Turner, J. Expression and hypoxic up-regulation of neuroglobin in human glioblastoma cells. Mol. Oncol 2009, 3, 45–53. [Google Scholar]
- Qin, H.; Guo, Y.; Zhang, C.; Zhang, L.; Li, M.; Guan, P. The expression of neuroglobin in astrocytoma. Brain Tumor. Pathol 2012, 29, 10–16. [Google Scholar]
- Mook-Jung, I.; Kim, H.; Fan, W.; Tezuka, Y.; Kadota, S.; Nishijo, H.; Jung, M.W. Neuroprotective effects of constituents of the oriental crude drugs, Rhodiola sacra, R. Sachalinensis and tokaku-joki-to, against β-amyloid toxicity, oxidative stress and apoptosis. Biol. Pharm. Bull 2002, 25, 1101–1104. [Google Scholar]
- Sawada, H.; Ibi, M.; Kihara, T.; Urushitani, M.; Akaike, A.; Shimohama, S. Estradiol protects mesencephalic dopaminergic neurons from oxidative stress-induced neuronal death. J. Neurosci. Res 1998, 54, 707–719. [Google Scholar]
- Green, P.S.; Simpkins, J.W. Neuroprotective effects of estrogens: Potential mechanisms of action. Int. J. Dev. Neurosci 2000, 18, 347–358. [Google Scholar]
- Wilson, M.E.; Dubal, D.B.; Wise, P.M. Estradiol protects against injury-induced cell death in cortical explant cultures: A role for estrogen receptors. Brain Res 2000, 873, 235–242. [Google Scholar]
- Behl, C.; Skutella, T.; Lezoualc’h, F.; Post, A.; Widmann, M.; Newton, C.J.; Holsboer, F. Neuroprotection against oxidative stress by estrogens: Structure-activity relationship. Mol. Pharmacol 1997, 51, 535–541. [Google Scholar]
- Gillies, G.E.; McArthur, S. Estrogen actions in the brain and the basis for differential action in men and women: A case for sex-specific medicines. Pharmacol. Rev 2010, 62, 155–198. [Google Scholar]
- Henderson, B.E.; Feigelson, H.S. Hormonal carcinogenesis. Carcinogenesis 2000, 21, 427–433. [Google Scholar]
- Key, T.J. Serum oestradiol and breast cancer risk. Endocr. Relat. Cancer 1999, 6, 175–180. [Google Scholar]
- Laviolette, L.A.; Garson, K.; Macdonald, E.A.; Senterman, M.K.; Courville, K.; Crane, C.A.; Vanderhyden, B.C. 17β-estradiol accelerates tumor onset and decreases survival in a transgenic mouse model of ovarian cancer. Endocrinology 2010, 151, 929–938. [Google Scholar]
- Bhat, H.K.; Calaf, G.; Hei, T.K.; Loya, T.; Vadgama, J.V. Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 3913–3918. [Google Scholar]
- Sittampalam, G.S.; Kahl, S.D.; Janzen, W.P. High-throughput screening: Advances in assay technologies. Curr. Opin. Chem. Biol 1997, 1, 384–391. [Google Scholar]
- Signore, A.P.; Zhang, F.; Weng, Z.; Gao, Y.; Chen, J. Leptin neuroprotection in the cns: Mechanisms and therapeutic potentials. J. Neurochem 2008, 106, 1977–1990. [Google Scholar]
- Ma, T.C.; Campana, A.; Lange, P.S.; Lee, H.H.; Banerjee, K.; Bryson, J.B.; Mahishi, L.; Alam, S.; Giger, R.J.; Barnes, S.; et al. A large-scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone daidzeinas a clinically approved small molecule that can promote neuronal protection or regeneration via a camp-independent pathway. J. Neurosci 2010, 30, 739–748. [Google Scholar]
- Ehrenreich, H.; Aust, C.; Krampe, H.; Jahn, H.; Jacob, S.; Herrmann, M.; Sirén, A.L. Erythropoietin: Novel approaches to neuroprotection in human brain disease. Metab. Brain Dis 2004, 19, 195–206. [Google Scholar]
- Fan, F.; Wood, K.V. Bioluminescent assays for high-throughput screening. Assay. Drug. Dev. Technol 2007, 5, 127–136. [Google Scholar]
- Zhang, W.; Tian, Z.; Sha, S.; Cheng, L.Y.; Philipsen, S.; Tan-Un, K.C. Functional and sequence analysis of human neuroglobin gene promoter region. Biochim. Biophys. Acta 2011, 1809, 236–244. [Google Scholar]
- Iljin, K.; Ketola, K.; Vainio, P.; Halonen, P.; Kohonen, P.; Fey, V.; Grafström, R.C.; Perälä, M.; Kallioniemi, O. High-throughput cell-based screening of 4910 known drugs and drug-like small molecules identifies disulfiram as an inhibitor of prostate cancer cell growth. Clin. Cancer Res 2009, 15. [Google Scholar] [CrossRef]
- Sink, R.; Gobec, S.; Pecar, S.; Zega, A. False positives in the early stages of drug discovery. Curr. Med. Chem 2010, 17, 4231–4255. [Google Scholar]
- Shun, T.Y.; Lazo, J.S.; Sharlow, E.R.; Johnston, P.A. Identifying actives from hts data sets: Practical approaches for the selection of an appropriate hts data-processing method and quality control review. J. Biomol. Screen 2011, 16, 1–14. [Google Scholar]
- Campbell, J.B. Improving lead generation success through integrated methods: Transcending “drug discovery by numbers”. IDrugs 2010, 13, 874–879. [Google Scholar]
- Guido, R.V.; Oliva, G.; Andricopulo, A.D. Modern drug discovery technologies: Opportunities and challenges in lead discovery. Comb. Chem. High Throughput Screen 2011, 14, 830–839. [Google Scholar]


© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yu, Z.; Liu, N.; Liu, J.; Yang, K.; Wang, X. Neuroglobin, a Novel Target for Endogenous Neuroprotection against Stroke and Neurodegenerative Disorders. Int. J. Mol. Sci. 2012, 13, 6995-7014. https://doi.org/10.3390/ijms13066995
Yu Z, Liu N, Liu J, Yang K, Wang X. Neuroglobin, a Novel Target for Endogenous Neuroprotection against Stroke and Neurodegenerative Disorders. International Journal of Molecular Sciences. 2012; 13(6):6995-7014. https://doi.org/10.3390/ijms13066995
Chicago/Turabian StyleYu, Zhanyang, Ning Liu, Jianxiang Liu, Kevin Yang, and Xiaoying Wang. 2012. "Neuroglobin, a Novel Target for Endogenous Neuroprotection against Stroke and Neurodegenerative Disorders" International Journal of Molecular Sciences 13, no. 6: 6995-7014. https://doi.org/10.3390/ijms13066995
APA StyleYu, Z., Liu, N., Liu, J., Yang, K., & Wang, X. (2012). Neuroglobin, a Novel Target for Endogenous Neuroprotection against Stroke and Neurodegenerative Disorders. International Journal of Molecular Sciences, 13(6), 6995-7014. https://doi.org/10.3390/ijms13066995