Spatial Simulations in Systems Biology: From Molecules to Cells

Abstract
:1. Introduction
2. The Mesoscale Level
2.1. Diffusion in the Cell
 (0,Δt); for convenience the Δt has been included in the square root in Equation (3) such that readily available standard normal random numbers can be used).
(0,Δt); for convenience the Δt has been included in the square root in Equation (3) such that readily available standard normal random numbers can be used).2.2. Reactions in the Cell
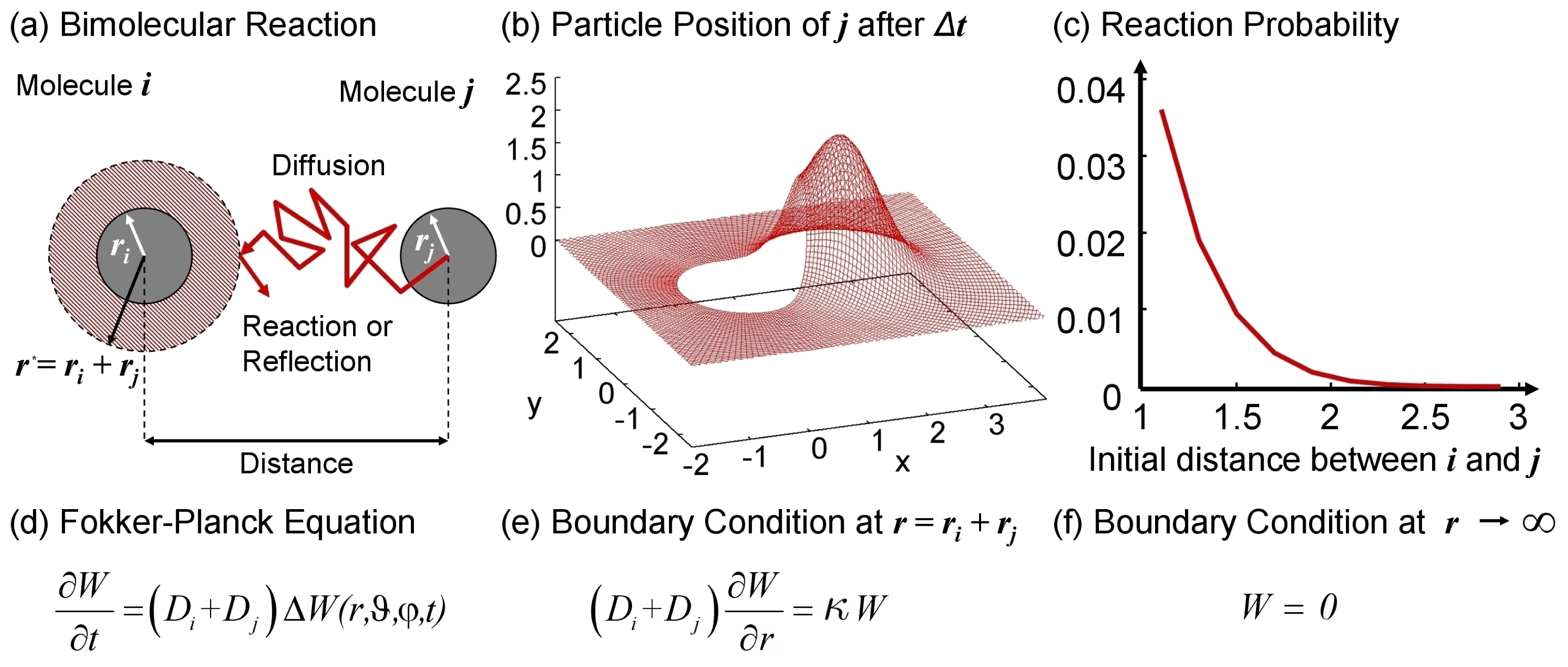
2.3. Reactions in the Simulation: Implementation Issues
 [0, 1] (and of course Pi < 1).
[0, 1] (and of course Pi < 1).- set the critical reaction radius to the physical collision radius
- and execute reactions for particles with ||xi(t) − xj(t)|| ≤ r*ij with probability
2.4. Performance and Accuracy
3. Applications and Results of Spatial Simulations
3.1. Current Spatial Simulation Frameworks for the Cellular Level
3.2. The Reaction Diffusion Master Equation and Gillespie’s Algorithm
 are the reaction and diffusion operators, respectively. The definition of the reaction operator follows from the classical master equation and thus reads reads(ν) denotes the volumes μ in the neighborhood of volume ν.
are the reaction and diffusion operators, respectively. The definition of the reaction operator follows from the classical master equation and thus reads reads(ν) denotes the volumes μ in the neighborhood of volume ν.3.3. Rule-based Modeling
3.4. Applications and Results
- Binding kinetics and binding sites: depending on the description level, protein-protein association can become quite complex [36]. For instance if multiple binding sites and diffusion-controlled reactions are considered. Biomolecules can have several binding sites for the same ligand, for instance receptors forming multimers or antibodies [128]. Kang et al. [173] analysed this and Park et al. [174] developed a theory for reversible reactions under these circumstances. For instance, two binding sites on a molecule would mean that the microscopic reaction rate constant κij is doubled, while the reaction radius is the same as for a molecule with just one binding site. Equation (6) shows that the macroscopic rate constant will not necessarily double under these circumstances.
- Scaffolds and Channeling: Both in signaling and metabolic pathways co-localization of related molecules has been observed. Obviously co-localization has advantages because the local high concentration boosts the reaction rate [4,80,106,175,176]. Specific and even nonspecific binding interactions which modify the localization properties of molecules can thus enhance reactions [177]. Note, that the localization requires that molecules do not diffuse around/away, such that there is a trade-off between advantages due to co-localization and disadvantages due to the reduced mobility [75,80].
- Protein DNA interactions: Transcription factors have to find their target site on the DNA amongst millions of binding sites, and they do it surprisingly efficiently, e.g., by combining 1D sliding and 3D diffusion [178]. For instance nonspecific interactions could enhance association rates respectively [177]. Note that even DNA is well organized in space [6]. The spatial organization of DNA strands plays an important role, but long DNA strands can obviously not be modeled with full atomic detail in a MDS simulation such that multi-scale approaches have to be employed [25]. The observed bursting kinetics of transcription rates is likewise explained using open and closed chromatin states, which involve large-scale transitions of the DNA state [179].
- Assembly and fusion processes: Large polymer structures such as the cytoskeleton filaments play an important role for the spatial organization of the cell. Guo et al. modeled the actin assembly using Brownian dynamics [180]. Langevin dynamics have been used to simulate the assembly of virus polymers [181]. A rule-based description was likewise used to analyze the emergence of complex structures [172]. Likewise the fusion of membrane enclosed structures like vesicles is important for the functionality of the cell [64,151,182]. Note that the interplay of cytoskeleton filaments, motor proteins and vesicles can enhance their fusion process [150], while the cytoskeleton structure is for instance organized by the aforementioned growth processes but also motors pulling them together and creating spatial patterns [68].
- Non-uniform molecule distributions in space: In order to grow/move in specific directions cells have to polarize into front and back, which is associated with nonsymmetric particle distributions across the cell [134,183]. In addition receptors on the membrane can cluster together [9,184], and the output of spatial simulations shows the importance of the spatial organization in the cell [9]. Note that again reversible binding and/or unspecific binding interactions influence these reaction rates [177,184].
4. Towards Multi-Scale Simulations from Atoms to Cells
Acknowledgments
References
- Tomita, M. Whole-cell simulation: A grand challenge of the 21st century. Trends Biotechnol 2001, 19, 205–210. [Google Scholar]
- Mathews, C.K. The cell-bag of enzymes or network of channels? J. Bacteriol 1993, 175, 6377–6381. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Ovádi, J.; Srere, P.A. Macromolecular compartmentation and channeling. Int. Rev. Cyt 1999, 192, 255–280. [Google Scholar]
- Bray, D. Signaling complexes: Biophysical constraints on intracellular communication. Ann. Rev. Biophys. Biomol. Struct 1998, 27, 59–75. [Google Scholar]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol 2006, 7, 165–176. [Google Scholar]
- Kholodenko, B.N.; Hancock, J.B.; Kolch, W. Signalling ballet in space and time. Nat. Rev. Mol. Cell Biol 2010, 11, 414–426. [Google Scholar]
- Costa, M.N.; Radhakrishnan, K.; Wilson, B.S.; Vlachos, D.G.; Edwards, J.S. Coupled stochastic spatial and non-spatial simulations of ErbB1 signaling pathways demonstrate the importance of spatial organization in signal transduction. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- ScienceVisuals. Available online: http://www.sciencevisuals.com accessed on 08 June 2012.
- De Heras Ciechomski, P.; Mange, R.; Peternier, A. Two-Phased Real-Time Rendering of Large Neuron Databases. Proceedings of the 2008 International Conference on Innovations in Information Technology, Al Ain, United Arab Emirates, 16–18 December 2008; pp. 712–716.
- Ando, T.; Skolnick, J. Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion. Proc. Natl. Acad. Sci. USA 2010, 107, 18457–18462. [Google Scholar]
- Rafelski, S.M.; Marshall, W.F. Building the cell: Design principles of cellular architecture. Nat. Rev. Mol. Cell Biol 2008, 9, 593–602. [Google Scholar]
- Bittig, A.T.; Uhrmacher, A.M. Spatial Modeling in Cell Biology at Multiple Levels. Proceedings of the Winter Simulation Conference (WSC), Baltimore MD, USA, 5–8 December 2010; pp. 608–619.
- Takahashi, K.; Arjunan, S.N.V.; Tomita, M. Space in systems biology of signaling pathways— Towards intracellular molecular crowding in silico. FEBS Lett 2005, 579, 1783–1788. [Google Scholar]
- Tolle, D.P.; le Novere, N. Particle-based stochastic simulation in systems biology. Curr. Bioinf 2006, 1, 315–320. [Google Scholar]
- Turner, T.E.; Schnell, S.; Burrage, K. Stochastic approaches for modelling in vivo reactions. Comput. Biol. Chem 2004, 28, 165–178. [Google Scholar]
- Ridgway, D.; Broderick, G.; Ellison, M.J. Accommodating space, time and randomness in network simulation. Curr. Opin. Biotechnol 2006, 17, 493–498. [Google Scholar]
- Burrage, K.; Burrage, P.; Leier, A.; Marquez-Lago, T.; Nicolau, D., Jr. Stochastic simulation for spatial modelling of dynamic process in a living cell. Des. Anal. Biomol. Circuits: Eng. Approaches Syst. Synth. Biol 2011, 43–62. [Google Scholar]
- Gillespie, D.T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comp. Phys 1976, 22, 403–434. [Google Scholar]
- Leach, A.R. Molecular Modelling: Principles and Applications; Pearson Education Ltd: Harlow, England, 2001. [Google Scholar]
- Schlick, T. Molecular Modeling and Simulation: An Interdisciplinary Guide; Springer Verlag: Berlin, Germany, 2010. [Google Scholar]
- Bionumbers. Available online: http://bionumbers.hms.harvard.edu/bionumber.aspx?&id=106198&ver=2 accessed on 8 June 2012.
- Tozzini, V. Coarse-grained models for proteins. Curr. Opin. Struct. Biol 2005, 15, 144–150. [Google Scholar]
- Villa, E.; Balaeff, A.; Mahadevan, L.; Schulten, K. Multiscale method for simulating protein-DNA complexes. Multiscale Model. Simul 2004, 2, 527–553. [Google Scholar]
- Chandran, P.L.; Mofrad, M.R.K. Averaged implicit hydrodynamic model of semiflexible filaments. Phys. Rev. E 2010, 81, 031920:1–031920:17. [Google Scholar]
- Cyron, C.J.; Wall, W.A. Numerical method for the simulation of the Brownian dynamics of rod-like microstructures with three-dimensional nonlinear beam elements. Int. J. Numer. Methods Eng 2012, 90, 955–987. [Google Scholar]
- Andrews, S.S.; Bray, D. Stochastic simulation of chemical reactions with spatial resolution and single molecule detail. Phys. Biol 2004, 1, 137–151. [Google Scholar]
- Sandersius, S.A.; Newman, T.J. Modeling cell rheology with the Subcellular Element Model. Phys. Biol 2008, 5. [Google Scholar] [CrossRef]
- Sbalzarini, I.F.; Walther, J.H.; Bergdorf, M.; Hieber, S.E.; Kotsalis, E.M.; Koumoutsakos, P. PPM–A highly efficient parallel particle–mesh library for the simulation of continuum systems. J. Comp. Phys 2006, 215, 566–588. [Google Scholar]
- Newman, T.J. Grid-free models of multicellular systems, with an application to large-scale vortices accompanying primitive streak formation. Curr. Topics Dev. Biol 2008, 81, 157–182. [Google Scholar]
- Holcombe, M.; Adra, S.; Bicak, M.; Chin, S.; Coakley, S.; Graham, A.; Green, J.; Greenough, C.; Jackson, D.; Kiran, M.; et al. Modelling complex biological systems using an agent-based approach. Integr. Biol 2012, 4, 53–64. [Google Scholar]
- Walker, D.C.; Southgate, J. The virtual cell a candidate co-ordinator for middle-outmodelling of biological systems. Brief. Bioinf 2009, 10, 450–461. [Google Scholar]
- Geyer, T. Many-particle Brownian and Langevin Dynamics Simulations with the Brownmove package. BMC Biophys 2011, 4. [Google Scholar] [CrossRef]
- Kim, T.; Hwang, W.; Lee, H.; Kamm, R.D. Computational analysis of viscoelastic properties of crosslinked actin networks. PLoS Comput. Biol 2009, 5. [Google Scholar] [CrossRef]
- Gabdoulline, R.R.; Wade, R.C. Protein-protein association: Investigation of factors influencing association rates by Brownian dynamics simulations. J. Mol. Biol 2001, 306, 1139–1155. [Google Scholar]
- Sun, J.; Weinstein, H. Toward realistic modeling of dynamic processes in cell signaling: Quantification of macromolecular crowding effects. J. Chem. Phys 2007, 127, 155105:1–155105:10. [Google Scholar]
- Schmidt, R.R.; Cifre, J.G.H.; de la Torre, J.G. Comparison of Brownian dynamics algorithms with hydrodynamic interaction. J. Chem. Phys 2011, 135, 084116:1–084116:10. [Google Scholar]
- Erban, R.; Chapman, S.J. Stochastic modelling of reaction–diffusion processes: Algorithms for bimolecular reactions. Phys. Biol 2009, 6, 046001. [Google Scholar]
- Weiss, M.; Elsner, M.; Kartberg, F.; Nilsson, T. Anomalous subdiffusion is a measure for cytoplasmic crowding in living cells. Biophys. J 2004, 87, 3518–3524. [Google Scholar]
- Ridgway, D.; Broderick, G.; Lopez-Campistrous, A.; Ru’aini, M.; Winter, P.; Hamilton, M.; Boulanger, P.; Kovalenko, A.; Ellison, M.J. Coarse-grained molecular simulation of diffusion and reaction kinetics in a crowded virtual cytoplasm. Biophys. J 2008, 94, 3748–3759. [Google Scholar]
- Klann, M.T.; Lapin, A.; Reuss, M. Stochastic simulation of signal transduction: Impact of the cellular architecture on diffusion. Biophys. J 2009, 96, 5122–5129. [Google Scholar]
- Trinh, S.; Arce, P.; Locke, B.R. Effective diffusivities of point-like molecules in isotropic porous media by monte carlo simulation. Trans. Porous Media 2000, 38, 241–259. [Google Scholar]
- Długosz, M.; Trylska, J. Diffusion in crowded biological environments: Applications of Brownian dynamics. BMC Biophys 2011, 4. [Google Scholar] [CrossRef]
- Chang, R.; Jagannathan, K.; Yethiraj, A. Diffusion of hard sphere fluids in disordered media: A molecular dynamics simulation study. Phys. Rev. E 2004, 69. [Google Scholar] [CrossRef]
- Ölveczky, B.P.; Verkman, A.S. Monte carlo analysis of obstructed diffusion in three dimensions: Application to molecular diffusion in organelles. Biophys. J 1998, 74, 2722–2730. [Google Scholar]
- Verkman, A.S. Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci 2002, 27, 27–33. [Google Scholar]
- Lipkow, K.; Andrews, S.S.; Bray, D. Simulated diffusion of phosphorylated CheY through the cytoplasm of Escherichia coli. J. Bacteriol 2005, 187, 45–53. [Google Scholar]
- Luby-Phelps, K. Cytoarchitecture and physical properties of cytoplasm: Volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol 2000, 192, 189–221. [Google Scholar]
- Jacobson, K.; Wojcieszyn, J. The translational mobility of substances within the cytoplasmic matrix. Proc. Natl. Acad. Sci. USA 1984, 81, 6747–6751. [Google Scholar]
- Blum, J.J.; Lawler, G.; Reed, M.; Shin, I. Effect of cytoskeletal geometry on intracellular diffusion. Biophys. J 1989, 56, 995–1005. [Google Scholar]
- Weissberg, H.L. Effective diffusion coefficient in porous media. J. Appl. Phys 1963, 34, 2636– 2639. [Google Scholar]
- Whitaker, S. The Method of Volume Averaging; Springer: Berlin, Germany, 1998. [Google Scholar]
- Fan, T.H.; Dhont, J.K.G.; Tuinier, R. Motion of a sphere through a polymer solution. Phys. Rev. E 2007, 75. [Google Scholar] [CrossRef]
- Ogston, A.G.; Preston, B.N.; Wells, J.D. On the transport of compact particles through solutions of chain-polymers. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci 1973, 333, 297–316. [Google Scholar]
- Cukier, R.I. Diffusion of Brownian spheres in semidilute polymer solutions. Macromolecules 1984, 17, 252–255. [Google Scholar]
- Han, J.; Herzfeld, J. Macromolecular diffusion in crowded solutions. Biophys. J 1993, 65, 1155–1161. [Google Scholar]
- Bruna, M.; Chapman, S.J. Excluded-volume effects in the diffusion of hard spheres. Phys. Rev. E 2012, 85. [Google Scholar] [CrossRef]
- Sakha, F.; Fazli, H. Three-dimensional Brownian diffusion of rod-like macromolecules in the presence of randomly distributed spherical obstacles: Molecular dynamics simulation. J. Chem. Phys 2010, 133, 234904:1–234904:6. [Google Scholar]
- Ando, T.; Skolnick, J. Brownian Dynamics Simulation of Macromolecule Diffusion in a Protocell. Proceedings of the International Conference of the Quantum Bio-Informatics IV, Tokyo, Japan, 10–13 March 2010; Georgia Institute of Technology and World Scientific Publishing: Atlanta, GA, USA, 2011; 28, pp. 413–426. [Google Scholar]
- Saxton, M.J. A biological interpretation of transient anomalous subdiffusion. I. Qualitative model. Biophys. J 2007, 92, 1178–1191. [Google Scholar]
- Hiroi, N.; Lu, J.; Iba, K.; Tabira, A.; Yamashita, S.; Okada, Y.; Flamm, C.; Oka, K.; Köhler, G.; Funahashi, A. Physiological environment induces quick response–slow exhaustion reactions. Frontiers Physiol 2011, 2. [Google Scholar] [CrossRef]
- Echeveria, C.; Tucci, K.; Kapral, R. Diffusion and reaction in crowded environments. J. Phys. Condens. Matter 2007, 19. [Google Scholar] [CrossRef]
- Shillcock, J. Insight or illusion? Seeing inside the cell with mesoscopic simulations. HFSP J 2008, 2, 1–6. [Google Scholar]
- Kapral, R. Multiparticle Collision Dynamics: Simulation of Complex Systems on Mesoscales. In Advances in Chemical Physics; Rice, S.A., Ed.; John Wiley and Sons, Inc: Hoboken, NJ, USA, 2008; Volume 140, pp. 89–146. [Google Scholar]
- Cyron, C.J.; Wall, W.A. Consistent finite-element approach to Brownian polymer dynamics with anisotropic friction. Phys. Rev. E 2010, 82, 66705:1–66705:12. [Google Scholar]
- Lee, H.; Pelz, B.; Ferrer, J.M.; Kim, T.; Lang, M.J.; Kamm, R.D. Cytoskeletal deformation at high strains and the role of cross-link unfolding or unbinding. Cell. Mol. Bioeng 2009, 2, 28–38. [Google Scholar]
- Karsenti, E.; Nédélec, F.; Surrey, T. Modelling microtubule patterns. Nat. Cell Biol 2006, 8, 1204–1211. [Google Scholar]
- Renkin, E.M. Multiple pathways of capillary permeability. Circ. Res 1977, 41, 735–743. [Google Scholar]
- Taylor, A.E.; Granger, D.N. Exchange of Macromolecules across the Microcirculation. In Handbook of Physiology: The Cardiovascular System: Microcirculation; American Physiological Society: Bethesda, MD, USA, 1984; Volume 4, pp. 467–520. [Google Scholar]
- Zimmerman, S.B.; Trach, S.O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol 1991, 222, 599–620. [Google Scholar]
- Niederalt, C. Bayer Technology Services. PK-Sim/MoBi from Bayer Technology Services. Personal communication, 2011. [Google Scholar]
- Vale, R.D. The molecular motor toolbox for intracellular transport. Cell 2003, 112, 467–480. [Google Scholar]
- Falk, M.; Klann, M.; Reuss, M.; Ertl, T. Visualization of Signal Transduction Processes in the Crowded Environment of the Cell. Proceedings of IEEE Pacific Visualization Symposium 2009 (PacificVis ’09), Beijing, China, 20–23 April 2009; pp. 169–176.
- Klann, M. Development of a Stochastic Multi-Scale Simulation Method for the Analysis of Spatiotemporal Dynamics in Cellular Transport and Signaling Processes. Ph.D. Dissertation, Universität Stuttgart, Germany, November 2011. [Google Scholar]
- Li, G.; Cui, Q. Mechanochemical coupling in myosin: A theoretical analysis with molecular dynamics and combined QM/MM reaction path calculations. J. Phys. Chem. B 2004, 108, 3342–3357. [Google Scholar]
- Kawakubo, T.; Okada, O.; Minami, T. Molecular dynamics simulations of evolved collective motions of atoms in the myosin motor domain upon perturbation of the ATPase pocket. Biophys. Chem 2005, 115, 77–85. [Google Scholar]
- Otten, M.; Nandi, A.; Arcizet, D.; Gorelashvili, M.; Lindner, B.; Heinrich, D. Local motion analysis reveals impact of the dynamic cytoskeleton on intracellular subdiffusion. Biophys. J 2012, 102, 758–767. [Google Scholar]
- Gershon, N.D.; Porter, K.R.; Trus, B.L. The cytoplasmic matrix: Its volume and surface area and the diffusion of molecules through it. Proc. Natl. Acad. Sci. USA 1985, 82, 5030–5034. [Google Scholar]
- Klann, M.T.; Lapin, A.; Reuss, M. Agent-based simulation of reactions in the crowded and structured intracellular environment: Influence of mobility and location of the reactants. BMC Syst. Biol 2011, 5. [Google Scholar] [CrossRef]
- Smoluchowski, M. Versuch einer mathematischen Theorie der Koagulationskinetik kolloider Lösungen. Z. Phys. Chem 1917, 92, 129–168. [Google Scholar]
- Zhang, Y.; McCammon, J.A. Studying the affinity and kinetics of molecular association with molecular-dynamics simulation. J. Chem. Phys 2003, 118, 1821:1–1821:7. [Google Scholar]
- Gabdoulline, R.R.; Wade, R.C. Biomolecular diffusional association. Curr. Opin. Struct. Biol 2002, 12, 204–213. [Google Scholar]
- Northrup, S.H.; Erickson, H.P. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. USA 1992, 89, 3338–3342. [Google Scholar]
- Rice, S.A. Diffusion-Limited Reactions; Elsevier: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Collins, F.C.; Kimball, G.E. Diffusion-controlled reaction rates. J. Colloid. Sci 1949, 4, 425– 437. [Google Scholar]
- Ellis, R.J. Macromolecular crowding: An important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol 2001, 11, 114–119. [Google Scholar]
- Minton, A.P. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem 2001, 276, 10577–10580. [Google Scholar]
- Al-Habori, M. Microcompartmentation, metabolic channelling and carbohydrate metabolism. Int. J. Biochem. Cell Biol 1995, 27, 123–132. [Google Scholar]
- Schnell, S.; Turner, T.E. Reaction kinetics in intracellular environments with macromolecular crowding: Simulations and rate laws. Prog. Biophys. Mol. Biol 2004, 85, 235–260. [Google Scholar]
- Grima, R.; Schnell, S. A systematic investigation of the rate laws valid in intracellular environments. Biophys. Chem 2006, 124, 1–10. [Google Scholar]
- Nicolau, D.V., Jr; Burrage, K. Stochastic simulation of chemical reactions in spatially complex media. Comput. Mathe. Appl 2008, 55, 1007–1018. [Google Scholar]
- Means, S.; Smith, A.J.; Shepherd, J.; Shadid, J.; Fowler, J.; Wojcikiewicz, R.J.H.; Mazel, T.; Smith, G.D.; Wilson, B.S. Reaction diffusion modeling of calcium dynamics with realistic ER geometry. Biophys. J 2006, 91, 537–557. [Google Scholar]
- Bergdorf, M.; Sbalzarini, I.F.; Koumoutsakos, P. A Lagrangian particle method for reaction–diffusion systems on deforming surfaces. J. Math. Biol 2010, 61, 649–663. [Google Scholar]
- Loverdo, C.; Benichou, O.; Moreau, M.; Voituriez, R. Enhanced reaction kinetics in biological cells. Nat. Phys 2008, 4, 134–137. [Google Scholar]
- Chaudhuri, A.; Bhattacharya, B.; Gowrishankar, K.; Mayor, S.; Rao, M. Spatiotemporal regulation of chemical reactions by active cytoskeletal remodeling. Proc. Natl. Acad. Sci. USA 2011, 108, 14825–14830. [Google Scholar]
- Hardt, S.L. Rates of diffusion controlled reactions in one, two and three dimensions. Biophys. Chem 1979, 10, 239–243. [Google Scholar]
- Torney, D.C.; McConnel, H.M. Diffusion-Limited reaction rate theory for two-dimensional systems. Proc. R. Soc. Lond. A 1983, 387, 147–170. [Google Scholar]
- Kholodenko, B.N.; Hoek, J.B.; Westerhoff, H.V. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol 2000, 10, 173–178. [Google Scholar]
- Lampoudi, S.; Gillespie, D.T.; Petzold, L.R. Effect of excluded volume on 2D discrete stochastic chemical kinetics. J. Comp. Phys 2009, 228, 3656–3668. [Google Scholar]
- Bisswanger, H. Enzyme Kinetics; Wiley VCH: Chichester, England, 2002. [Google Scholar]
- Rohwer, J.; Hanekom, A.; Hofmeyr, J.H. A Universal Rate Equation for Systems Biology. Proceedings of the 2nd International Symposium on Experimental Standard Conditions of Enzyme Characterizations (ESEC 2006), Rüdesheim, Germany, 19–23 March 2006; pp. 175–187.
- Segel, L.A.; Slemrod, M. The quasi-steady-state assumption: A case study in perturbation. SIAM Rev 1989, 31, 446–477. [Google Scholar]
- Byrne, M.J.; Waxham, M.N.; Kubota, Y. Cellular dynamic simulator: An event driven molecular simulation environment for cellular physiology. Neuroinformatics 2010, 8, 63–82. [Google Scholar]
- Arjunan, S.N.V.; Tomita, M. A new multicompartmental reaction-diffusion modeling method links transient membrane attachment of E. coli MinE to E-ring formation. M. Syst. Synth. Biol 2010, 4, 35–53. [Google Scholar]
- Klann, M.; Ganguly, A.; Koeppl, H. Improved Reaction Scheme for Spatial Stochastic Simulations with Single Molecule Detail. Proceedings of the 8th International Workshop on Computational Systems Biology (WCSB 2011), Zurich, Switzerland, 6–8 June 2011.
- Falk, M.; Klann, M.; Ott, M.; Ertl, T.; Koeppl, H. Parallelized Agent-Based Simulation on CPU and Graphics Hardware for Spatial and Stochastic Models in Biology. Proceedings of the 9th International Conference on Computational Methods in Systems Biology, Paris, France, 21–23 September 2011; pp. 73–82.
- Clifford, P.; Green, N.J.B. On the simulation of the Smoluchowski boundary condition and the interpolation of brownian paths. Mol. Phys 1986, 57, 123–128. [Google Scholar]
- Lapin, A.; Klann, M.; Reuss, M. Stochastic Simulations of 4D Spatial Temporal Dynamics of Signal Transduction Processes. Proceedings of the FOSBE, Stuttgart, Germany, 9–12 September 2007; pp. 421–425.
- Van Zon, J.S.; ten Wolde, P.R. Simulating biochemical networks at the particle level and in time and space: Green’s function reaction dynamics. Phys. Rev Lett 2005, 94. [Google Scholar] [CrossRef]
- Morelli, M.J.; ten Wolde, P.R. Reaction Brownian dynamics and the effect of spatial fluctuations on the gain of a push-pull network. J. Chem. Phys 2008, 129. [Google Scholar] [CrossRef]
- Kim, H.; Shin, K.J. Exact solution of the reversible diffusion-influenced reaction for an isolated pair in three dimensions. Phys. Rev. Lett 1999, 82, 1578–1581. [Google Scholar]
- Gopich, I.V.; Szabo, A. Kinetics of reversible diffusion influenced reactions: The self-consistent relaxation time approximation. J. Chem. Phys 2002, 117, 507:1–507:11. [Google Scholar]
- Lapin, A.; Klann, M.; Reuss, M. Multi-Scale Spatio-Temporal Modeling: Lifelines of Microorganisms in Bioreactors and Tracking Molecules in Cells. In Biosystems Engineering II; Springer-Verlag: Berlin, Germany, 2010; Volume 121, pp. 23–43. [Google Scholar]
- Park, S.; Agmon, N. Theory and simulation of diffusion-controlled michaelis-menten kinetics for a static enzyme in solution. J. Phys. Chem. B 2008, 112, 5977–5987. [Google Scholar]
- Pogson, M.; Smallwood, R.; Qwarnstrom, E.; Holcombe, M. Formal agent-based modelling of intracellular chemical interactions. Biosystems 2006, 85, 37–45. [Google Scholar]
- Pogson, M.; Holcombe, M.; Smallwood, R.; Qwarnstrom, E. Introducing spatial information into predictive NF-κB modelling—An agent-based approach. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Lipková, J.; Zygalakis, K.C.; Chapman, S.J.; Erban, R. Analysis of Brownian dynamics simulations of reversible bimolecular reactions. SIAM J. Appl. Math 2011, 71, 714–730. [Google Scholar]
- Andrews, S.S.; Addy, N.J.; Brent, R.; Arkin, A.P. Detailed simulations of cell biology with Smoldyn 2.1. PLoS Comp. Biol 2010, 6. [Google Scholar] [CrossRef]
- Andrews, S.S. Serial rebinding of ligands to clustered receptors as exemplified by bacterial chemotaxis. Phys. Biol 2005, 2, 111–122. [Google Scholar]
- Berg, O.G. On diffusion-controlled dissociation. Chem. Phys 1978, 31, 47–57. [Google Scholar]
- Klann, M.; Koeppl, H. Escape times and geminate recombinations in spatial simulations of chemical reactions. Biophys. J 2012.
- Wade, R.C.; Luty, B.A.; Demchuk, E.; Madura, J.D.; Davis, M.E.; Briggs, J.M.; McCammon, J.A. Simulation of enzyme–substrate encounter with gated active sites. Nat. Struct. Mol. Biol 1994, 1, 65–69. [Google Scholar]
- Shoup, D.; Lipari, G.; Szabo, A. Diffusion-controlled bimolecular reaction rates. The effect of rotational diffusion and orientation constraints. Biophys. J 1981, 36, 697–714. [Google Scholar]
- Dudko, O.K.; Berezhkovskii, A.M.; Weiss, G.H. Rate constant for diffusion-influenced ligand binding to receptors of arbitrary shape on a cell surface. J. Chem. Phys 2004, 121, 1562–1565. [Google Scholar]
- Traytak, S.D. Diffusion-controlled reaction rate to an active site. Chem. Phys 1995, 192, 1–7. [Google Scholar]
- Wu, Y.T.; Nitsche, J.M. On diffusion-limited site-specific association processes for spherical and nonspherical molecules. Chem. Eng. Sci 1995, 50, 1467–1487. [Google Scholar]
- Bongini, L.; Fanelli, D.; Piazza, F.; de los Rios, P.; Sanner, M.; Skoglund, U. A dynamical study of antibody–antigen encounter reactions. Phys. Biol 2007, 4, 172–180. [Google Scholar]
- Pettré, J.; Ciechomski, P.H.; Maïm, J.; Yersin, B.; Laumond, J.P.; Thalmann, D. Real-time navigating crowds: Scalable simulation and rendering. Comput. Animat. Virtual Worlds 2006, 17, 445–455. [Google Scholar]
- Behringer, H.; Eichhorn, R. Hard-wall interactions in soft matter systems: Exact numerical treatment. Phys. Rev. E 2011, 83, 065701:1–065701:4. [Google Scholar]
- Press, W.H.; Flannery, B.P.; Teukolsky, S.A.; Vetterling, W.T. Numerical Recipes; Cambridge University Press: Cambridge, UK, 1986; Volume 547. [Google Scholar]
- Trotter, H.F. An elementary proof of the central limit theorem. Arch. Math 1959, 10, 226–234. [Google Scholar]
- Dematte, L. Smoldyn on graphics processing units: Massively parallel brownian dynamics simulation. IEEE/ACM Trans. Comput. Biol. Bioinf 2012, 9, 655–667. [Google Scholar]
- Jilkine, A.; Angenent, S.B.; Wu, L.F.; Altschuler, S.J. A density-dependent switch drives stochastic clustering and polarization of signaling molecules. PLoS Comp. Biol 2011, 7. [Google Scholar] [CrossRef]
- Plimpton, S.; Slepoy, A. ChemCell: A Particle-Based Model of Protein Chemistry and Diffusion in Microbial Cells; Sandia National Laboratories Technical Report 2003-4509; Sandia National Laboratories: Albuquerque, USA, 2003. [Google Scholar]
- Plimpton, S.J.; Slepoy, A. Microbial cell modeling via reacting diffusive particles. J. Phys. Conf. Ser 2005, 16. [Google Scholar] [CrossRef]
- Takahashi, K.; Ishikawa, N.; Sadamoto, Y.; Sasamoto, H.; Ohta, S.; Shiozawa, A.; Miyoshi, F.; Naito, Y.; Nakayama, Y.; Tomita, M. E-Cell 2: Multi-platform E-Cell simulation system. Bioinformatics 2003, 19, 1727–1729. [Google Scholar]
- Tomita, M.; Hashimoto, K.; Takahashi, K.; Shimizu, T.S.; Matsuzaki, Y.; Miyoshi, F.; Saito, K.; Tanida, S.; Yugi, K.; Venter, J.C.; et al. E-CELL: Software environment for whole-cell simulation. Bioinformatics 1999, 15, 72–84. [Google Scholar]
- Takahashi, K.; Kaizu, K.; Hu, B.; Tomita, M. A multi-algorithm, multi-timescale method for cell simulation. Bioinformatics 2004, 20, 538–546. [Google Scholar]
- Takahashi, K.; Tănase-Nicola, S.; ten Wolde, P.R. Spatio-temporal correlations can drastically change the response of a MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 2473–2478. [Google Scholar]
- Van Zon, J.S.; Morelli, M.J.; Tanase-Nicola, S.; tenWolde, P.R. Diffusion of transcription factors can drastically enhance the noise in gene expression. Biophys. J 2006, 91, 4350–4367. [Google Scholar]
- Stiles, J.R.; Bart, T.M. Monte Carlo Methods for Simulating Realistic Synaptic Microphysiology Using MCell. In Computational Neuroscience—Realistic Modeling for Experimentalists; de Schutter, E., Ed.; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Hattne, J.; Fange, D.; Elf, J. Stochastic reaction-diffusion simulation with MesoRD. Bioinformatics 2005, 21, 2923–2924. [Google Scholar]
- Elf, J.; Doncic, A.; Ehrenberg, M. Mesoscopic reaction-diffusion in intracellular signaling. Proc. SPIE 2003, 5110, 114–124. [Google Scholar]
- Ander, M.; Beltrao, P.; di Ventura, B.; Ferkinghoff-Borg, J.; Foglierini, M.; Kaplan, A.; Lemerle, C.; Tomas-Oliveira, I.; Serrano, L. SmartCell, a framework to simulate cellular processes that combines stochastic approximation with diffusion and localisation: Analysis of simple networks. Syst. Biol 2004, 1, 129–138. [Google Scholar]
- Wils, S.; de Schutter, E. STEPS: Modeling and simulating complex reaction-diffusion systems with Python. Frontiers Neuroinf 2009, 3. [Google Scholar] [CrossRef]
- Stoma, S.; Fröhlich, M.; Gerber, S.; Klipp, E. STSE: Spatio-temporal simulation environment dedicated to biology. BMC Bioinf 2011, 12. [Google Scholar] [CrossRef]
- Moraru, I.I.; Schaff, J.C.; Slepchenko, B.M.; Loew, L.M. The virtual cell. Ann. N. Y. Acad. Sci 2002, 971, 595–596. [Google Scholar]
- Slepchenko, B.M.; Schaff, J.C.; Macara, I.; Loew, L.M. Quantitative cell biology with the virtual cell. Trends Cell Biol 2003, 13, 570–576. [Google Scholar]
- Klann, M.; Koeppl, H.; Reuss, M. Spatial modeling of vesicle transport and the cytoskeleton: The challenge of hitting the right road. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Shillcock, J.C.; Lipowsky, R. The computational route from bilayer membranes to vesicle fusion. J. Phys. Condens. Matt 2006, 18. [Google Scholar] [CrossRef]
- Liou, S.H.; Chen, C.C. Cellular ability to sense spatial gradients in the presence of multiple competitive ligands. Phys. Rev. E 2012, 85, 011904:1–011904:5. [Google Scholar]
- Angermann, B.R.; Klauschen, F.; Garcia, A.D.; Prustel, T.; Zhang, F.; Germain, R.N.; Meier-Schellersheim, M. Computational modeling of cellular signaling processes embedded into dynamic spatial contexts. Nat. Methods 2012, 9, 283–289. [Google Scholar]
- Jeschke, M.; Uhrmacher, A.M. Multi-Resolution Spatial Simulation for Molecular Crowding. Proceedings of the 2008 Winter Simulation Conference, Miami, FL, USA, 7–10 December 2008; pp. 1384–1392.
- Pahle, J. Biochemical simulations: Stochastic, approximate stochastic and hybrid approaches. Brief. Bioinf 2009, 10, 53–64. [Google Scholar]
- Chatterjee, A.; Vlachos, D.G. Multiscale spatial monte carlo simulations: Multigriding, computational singular perturbation, and hierarchical stochastic closures. J. Chem. Phys 2006, 124, 064110. [Google Scholar]
- Jeschke, M.; Ewald, R.; Park, A.; Fujimoto, R.; Uhrmacher, A.M. A parallel and distributed discrete event approach for spatial cell-biological simulations. ACM SIGMETRICS Perform. Eval. Rev 2008, 35, 22–31. [Google Scholar]
- Xing, F.; Yao, Y.P.; Jiang, Z.W.; Wang, B. Fine-grained parallel and distributed spatial stochastic simulation of biological reactions. Adv. Mater. Res 2012, 345, 104–112. [Google Scholar]
- Rodríguez, J.V.; Kaandorp, J.A.; Dobrzyński, M.; Blom, J.G. Spatial stochastic modelling of the phosphoenolpyruvate-dependent phosphotransferase (PTS) pathway in Escherichia coli. Bioinformatics 2006, 22, 1895–1901. [Google Scholar]
- Lampoudi, S.; Gillespie, D.T.; Petzold, L.R. The multinomial simulation algorithm for discrete stochastic simulation of reaction-diffusion systems. J. Chem. Phys 2009, 130, 094104. [Google Scholar]
- Stundzia, A.B.; Lumsden, C.J. Stochastic simulation of coupled reaction-diffusion processes. J. Comp. Phys 1996, 127, 196–207. [Google Scholar]
- Anderson, D.F. A modified next reaction method for simulating chemical systems with time dependent propensities and delays. J. Chem. Phys 2007, 127, 214107:1–214107:10. [Google Scholar]
- Gillespie, D.T. A diffusional bimolecular propensity function. J. Chem. Phys 2009, 131, 164109:1–164109:13. [Google Scholar]
- Fange, D.; Berg, O.G.; Sjöberg, P.; Elf, J. Stochastic reaction-diffusion kinetics in the microscopic limit. Proc. Natl. Acad. Sci. USA 2010, 107, 19820–19825. [Google Scholar]
- Gillespie, D.T. Deterministic limit of stochastic chemical kinetics. J. Phys. Chem. B 2009, 113, 1640–1644. [Google Scholar]
- Danos, V.; Feret, J.; Fontana, W.; Harmer, R.; Krivine, J. Rule-based Modelling of Cellular Signalling. Proceedings of the Eighteenth International Conference on Concurrency Theory, CONCUR’07, Lisbon, Portugal, 3–8 September 2007, Caires, L., Vasconcelos, V.T., Eds.; Springer: Berlin, Germany, 2007; Volume 4703, pp. 17–41. [Google Scholar]
- Faeder, J.R.; Blinov, M.L.; Hlavacek, W.S. Rule-based modeling of biochemical systems with BioNetGen. Methods Mol. Biol 2009, 500, 113–167. [Google Scholar]
- Camporesi, F.; Feret, J.; Koeppl, H.; Petrov, T. Automatic Reduction of Stochastic Rules-Based Models in a Nutshell. AIP Conference Proceedings. International Conference of Numerical Analysis and Applied Mathematics (ICNAAM 2010), Rhodes, Greece, 19–25 September 2010; 1281, pp. 1330–1334.
- Petrov, T.; Ganguly, A.; Koeppl, H. Model decomposition and stochastic fragments. Theor. Comput. Sci 2012, (in press). [Google Scholar]
- Tolle, D.P.; le Novère, N. Meredys, a multi-compartment reaction-diffusion simulator using multistate realistic molecular complexes. BMC Syst. Biol 2010, 4. [Google Scholar] [CrossRef]
- Yang, J.; Meng, X.; Hlavacek, W.S. Rule-based modelling and simulation of biochemical systems with molecular finite automata. Syst. Biol. IET 2010, 4, 453–466. [Google Scholar]
- Gruenert, G.; Ibrahim, B.; Lenser, T.; Lohel, M.; Hinze, T.; Dittrich, P. Rule-based spatial modeling with diffusing, geometrically constrained molecules. BMC Bioinf 2010, 11. [Google Scholar] [CrossRef]
- Kang, A.; Kim, J.H.; Lee, S.; Park, H. Diffusion-influenced reactions involving a reactant with two active sites. J. Chem. Phys 2009, 130, 094507:1–094507:8. [Google Scholar]
- Park, S.; Agmon, N. Multisite reversible geminate reaction. J. Chem. Phys 2009, 130, 074507:1–074507:11. [Google Scholar]
- Bauler, P.; Huber, G.; Leyh, T.; McCammon, J.A. Channeling by proximity: The catalytic advantages of active site colocalization using Brownian dynamics. J. Phys. Chem. Lett 2010, 1, 1332–1335. [Google Scholar]
- Locasale, J.W.; Shaw, A.S.; Chakraborty, A.K. Scaffold proteins confer diverse regulatory properties to protein kinase cascades. Proc. Natl. Acad. Sci. USA 2007, 104. [Google Scholar] [CrossRef]
- Zhou, H.X.; Szabo, A. Enhancement of association rates by nonspecific binding to DNA and cell membranes. Phys. Rev. Lett. 2004, 93, 178101:1–178101:4. [Google Scholar]
- Halford, S.E. An end to 40 years of mistakes in DNA-protein association kinetics? Biochem. Soc. Trans 2009, 37, 343–348. [Google Scholar]
- Zechner, C.; Ruess, J.; Krenn, P.; Pelet, S.; Peter, M.; Lygeros, J.; Koeppl, H. Moment-based inference predicts bimodality in transient gene expression. Proc. Natl. Acad. Sci. USA 2012, (in press). [Google Scholar]
- Guo, K.; Shillcock, J.; Lipowsky, R. Treadmilling of actin filaments via Brownian dynamics simulations. J. Chem. Phys 2010, 133. [Google Scholar] [CrossRef]
- Mahalik, J.P.; Muthukumar, M. Langevin dynamics simulation of polymer-assisted virus-like assembly. J. Chem. Phys 2012, 136, 135101:1–135101:13. [Google Scholar]
- Noguchi, H.; Takasu, M. Fusion pathways of vesicles: A Brownian dynamics simulation. J. Chem. Phys 2001, 115, 9547–9551. [Google Scholar]
- Mogilner, A.; Allard, J.; Wollman, R. Cell polarity: Quantitative modeling as a tool in cell biology. Science 2012, 336, 175–179. [Google Scholar]
- Mugler, A.; Bailey, A.G.; Takahashi, K.; Wolde, P.R. Membrane clustering and the role of rebinding in biochemical signaling. Biophys. J 2012, 102, 1069–1078. [Google Scholar]
- Cichocki, B.; Ekiel-Jezewska, M.L.; Wajnryb, E. Communication: Translational Brownian motion for particles of arbitrary shape. J. Chem. Phys 2012, 136, 071102:1–071102:4. [Google Scholar]
- Lee, D.; Redfern, O.; Orengo, C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol 2007, 8, 995–1005. [Google Scholar]
- Zhou, H.; Skolnick, J. Protein structure prediction by pro-Sp3-TASSER. Biophys. J 2009, 96, 2119–2127. [Google Scholar]
- Accelrys Discovery Studio. Available online: http://accelrys.com/products/discovery-studio/index.html accessed on 8 June 2012.
- Molsoft. Available online: http://www.molsoft.com accessed on 8 June 2012.
- García de la Torre, J.; Huertas, M.L.; Carrasco, B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J 2000, 78, 719–730. [Google Scholar]
- Moal, I.H.; Bates, P.A. Kinetic rate constant prediction supports the conformational selection mechanism of protein binding. PLoS Comp. Biol 2012, 8. [Google Scholar] [CrossRef]
- De Jong, D.H.; Schäfer, L.V.; de Vries, A.H.; Marrink, S.J.; Berendsen, H.J.C.; Grubmüller, H. Determining equilibrium constants for dimerization reactions from molecular dynamics simulations. J. Comp. Chem 2011, 32. [Google Scholar] [CrossRef]
- Lee, J.; Yang, S.; Kim, J.; Lee, S. An efficient molecular dynamics simulation method for calculating the diffusion-influenced reaction rates. J. Chem. Phys 2004, 120, 7564–7575. [Google Scholar]
- Thomas, A.S.; Elcock, A.H. Direct measurement of the kinetics and thermodynamics of association of hydrophobic molecules from molecular dynamics simulations. J. Phys. Chem. Lett 2011, 2, 19–24. [Google Scholar]
- Thomas, A.S.; Elcock, A.H. Direct observation of salt effects on molecular interactions through explicit-solvent molecular dynamics simulations: Differential effects on electrostatic and hydrophobic interactions and comparisons to Poisson-Boltzmann theory. J. Am. Chem. Soc 2006, 128, 7796–7806. [Google Scholar]
- Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D.G. How enzymes work: Analysis by modern rate theory and computer simulations. Science 2004, 303, 186–195. [Google Scholar]
- Gabdoulline, R.R.; Wade, R.C. Simulation of the diffusional association of barnase and barstar. Biophys. J 1997, 72, 1917–1929. [Google Scholar]
- Schlosshauer, M.; Baker, D. Realistic protein–protein association rates from a simple diffusional model neglecting long-range interactions, free energy barriers, and landscape ruggedness. Protein Sci 2004, 13, 1660–1669. [Google Scholar]
- Peter, E.; Dick, B.; Baeurle, S.A. A novel computer simulation method for simulating the multiscale transduction dynamics of signal proteins. J. Chem. Phys 2012, 136, 124112:1–124112:14. [Google Scholar]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar]
- Fujioka, A.; Terai, K.; Itoh, R.E.; Aoki, K.; Nakamura, T.; Kuroda, S.; Nishida, E.; Matsuda, M. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J. Biol. Chem 2006, 281, 8917–8926. [Google Scholar]
- Gutenkunst, R.N.; Waterfall, J.J.; Casey, F.P.; Brown, K.S.; Myers, C.R.; Sethna, J.P. Universally sloppy parameter sensitivities in systems biology models. PLoS Comp. Biol 2007, 3. [Google Scholar] [CrossRef]
- Chen, W.W.; Schoeberl, B.; Jasper, P.J.; Niepel, M.; Nielsen, U.B.; Lauffenburger, D.A.; Sorger, P.K. Input–output behavior of ErbB signaling pathways as revealed by a mass action model trained against dynamic data. Mol. Syst. Biol 2009, 5. [Google Scholar] [CrossRef]
- Hengl, S.; Kreutz, C.; Timmer, J.; Maiwald, T. Data-based identifiability analysis of non-linear dynamical models. Bioinformatics 2007, 23, 2612–2618. [Google Scholar]
- Koh, G.; Hsu, D.; Thiagarajan, P.S. Component-based construction of bio-pathway models: The parameter estimation problem. Theor. Comput. Sci 2011. [Google Scholar] [CrossRef]
- Gopalakrishnan, M.; Forsten-Williams, K.; Nugent, M.A.; Täuber, U.C. Effects of receptor clustering on ligand dissociation kinetics: Theory and simulations. Biophys. J 2005, 89, 3686–3700. [Google Scholar]
- Falk, M.; Klann, M.; Reuss, M.; Ertl, T. 3D Visualization of Concentrations from Stochastic Agent-based Signal Transduction Simulations. Proceedings of the IEEE International Symposium on Biomedical Imaging: From Nano to Macro, 2010, Rotterdam, The Netherlands, 14–17 April 2010; pp. 1301–1304.



{kind=link}
{kind=link}
{kind=link}
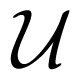{kind=link}
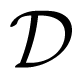{kind=link}
| Hydrodynamic Radius [nm] | Reference | |
|---|---|---|
| (i) | rh = 0.6169 × MW0.4226 | [72] |
| (ii) | rh = 0.7468 × MW1/3 | [41] |
| (iii) | rh = 0.5429 × MW1/3 | [40] |
| Name/Authors | Features | Website/References |
|---|---|---|
| Smoldyn S. Andrews et al. | Particle based simulator for reaction diffusion processes in arbitrarily shaped compartments. (point particles, no crowding). | www.smoldyn.org [28,48,119,120,133,134] |
| ChemCell | Particle based simulator within realistic cell shapes. | chemcell.sandia.gov [93,135,136] |
| E-Cell | Complete software environment for simulations on several levels. Contains further analysis tools. | www.e-cell.org [137–139] |
| (GFRD,eGFRD) ten Wolde et al. | Green’s function reaction dynamics will be included in the E-Cell project | [110,111,140,141] |
| FLAME | Agent-based multi-scale simulation (also beyond the cellular level). | www.flame.ac.uk [32,116,117] |
| MCell | Monte Carlo simulator of reaction diffusion processes. Reactions can only happen at membranes | www.mcell.cnl.salk.edu [142] |
| MesoRD | Spatial derivative of Gillespie’s algorithm to solve the Reaction-Diffusion Master Equation (RDME) with the “next subvolume method” | mesord.sourceforge.net [143,144] |
| SmartCell Serrano et al. | Spatial derivative of Gillespie’s algorithm in arbitrarily shaped compartments. | software.crg.es/smartcell [145] |
| STEPS | Tetrahedral mesh based spatial derivative of Gillespie’s algorithm | steps.sourceforge.net/STEPS [146] |
| STSE S.Stoma | PDE based simulator with compartments and direct linking to microscope images. | www.stse-software.org [147] |
| V Cell | ODE/PDE or SDE based simulator within realistic cell shapes. | www.nrcam.uchc.edu [148,149] |
| M. Klann et al. | Agent-based Brownian dynamics including cytoskeleton, crowding and vesicle transport. | www.bison.ethz.ch/research/spatial simulations [75,80,106,107,122,150] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Klann, M.; Koeppl, H. Spatial Simulations in Systems Biology: From Molecules to Cells. Int. J. Mol. Sci. 2012, 13, 7798-7827. https://doi.org/10.3390/ijms13067798
Klann M, Koeppl H. Spatial Simulations in Systems Biology: From Molecules to Cells. International Journal of Molecular Sciences. 2012; 13(6):7798-7827. https://doi.org/10.3390/ijms13067798
Chicago/Turabian StyleKlann, Michael, and Heinz Koeppl. 2012. "Spatial Simulations in Systems Biology: From Molecules to Cells" International Journal of Molecular Sciences 13, no. 6: 7798-7827. https://doi.org/10.3390/ijms13067798
APA StyleKlann, M., & Koeppl, H. (2012). Spatial Simulations in Systems Biology: From Molecules to Cells. International Journal of Molecular Sciences, 13(6), 7798-7827. https://doi.org/10.3390/ijms13067798