Possible Alterations in β-Synuclein, the Non-Amyloidogenic Homologue of α-Synuclein, during Progression of Sporadic α-Synucleinopathies

{kind=link}
Abstract
:1. Introduction
2. βS with a DLB-Linked Mutation Shows “Gain of Function” in α-Synucleinopathies
3. Does “Loss of Function” Explain the Role of Wild Type βS in Sporadic α-Synucleinopathies?
4. Rebutting Genetic Reports Showing a Negative Association of Wild Type βS with Sporadic α-Synucleinopathies
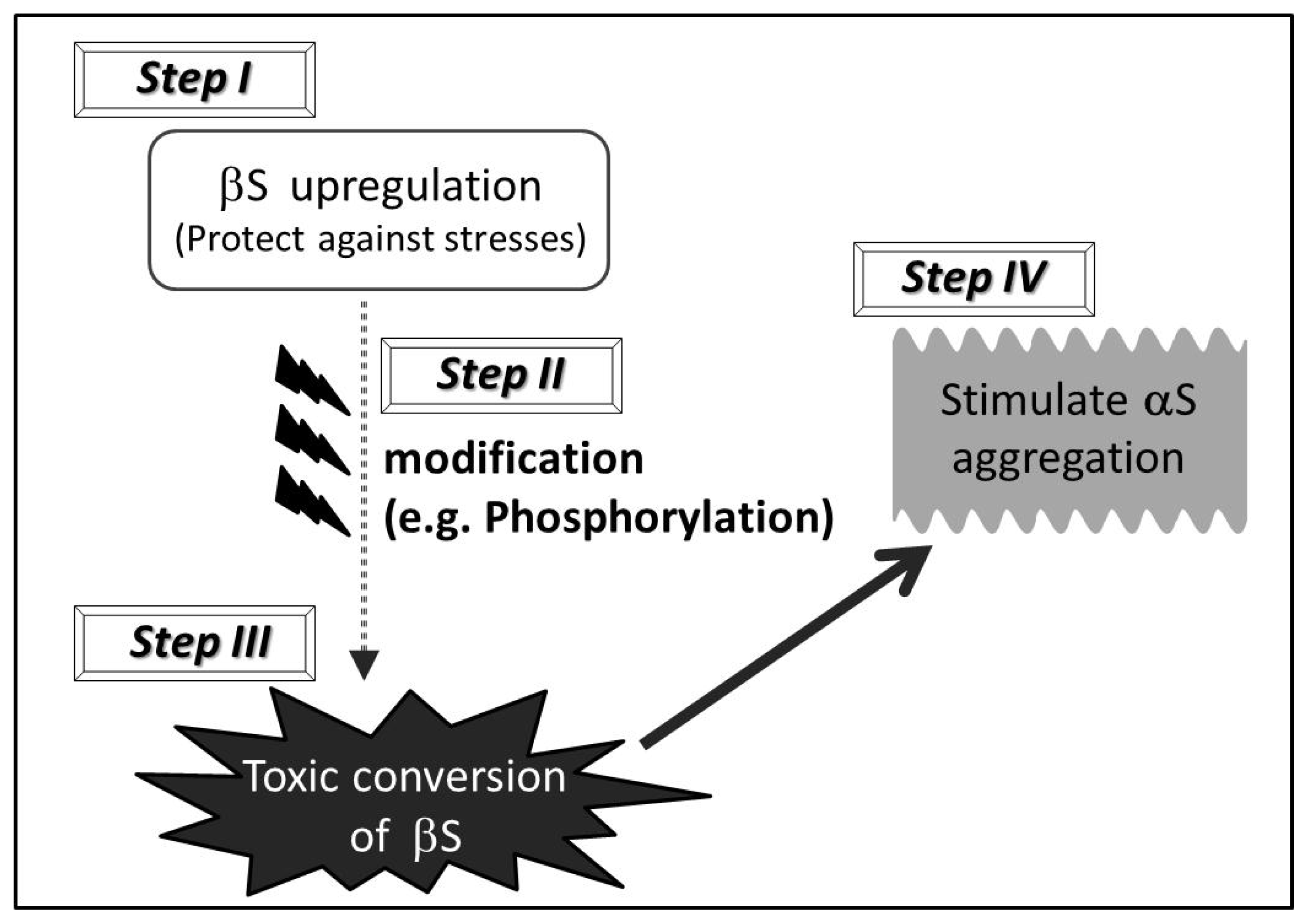
5. “Toxic Gain of Function” Scenario of Wild Type βS
6. Evidence that Wild Type βS Exhibits “Gain of Function” Properties in α-Synucleinopathies
7. Towards Novel Therapeutic Strategies
8. Conclusion
Acknowledgments
References
- Nakajo, S.; Tsukada, K.; Omata, K.; Nakamura, Y.; Nakaya, K. A new brain-specific 14-kDa protein is a phosphoprotein. Its complete amino acid sequence and evidence for phosphorylation. Eur. J. Biochem 1993, 217, 1057–1063. [Google Scholar]
- Fujita, M.; Hashimoto, M. Perspective on the Role of β-Synuclein in the Pathogenesis of α-Synucleinopathies. In Neuropathology: New Research; Almeirão, E., Honrado, T., Eds.; Nova Science Publishers: New York, NY, USA, 2012. [Google Scholar]
- Hashimoto, M.; La Spada, A.R. β-synuclein in the pathogenesis of Parkinson’s disease and related α-synucleinopathies: Emarging roles and new directions. Future Neurol 2012, 7, 155–163. [Google Scholar]
- Hashimoto, M.; Masliah, E. Alpha-synuclein in Lewy body disease and Alzheimer’s disease. Brain Pathol 1999, 9, 707–720. [Google Scholar]
- Trojanowski, J.Q.; Goedert, M.; Iwatsubo, T.; Lee, V.M. Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Differ 1998, 5, 832–837. [Google Scholar]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci 1998, 21, 249–254. [Google Scholar]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. Beta-synuclein inhibits alpha-synuclein aggregation: A possible role as an anti-parkinsonian factor. Neuron 2001, 32, 213–223. [Google Scholar]
- Fujita, M.; Sugama, S.; Sekiyama, K.; Sekigawa, A.; Tsukui, T.; Nakai, M.; Waragai, M.; Takenouchi, T.; Takamatsu, Y.; Wei, J.; et al. A β-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat. Commun 2010. [Google Scholar] [CrossRef]
- Wei, J.; Fujita, M.; Nakai, M.; Waragai, M.; Watabe, K.; Akatsu, H.; Rockenstein, E.; Masliah, E.; Hashimoto, M. Enhanced lysosomal pathology caused by beta-synuclein mutants linked to dementia with Lewy bodies. J. Biol. Chem 2007, 282, 28904–28914. [Google Scholar]
- Rockenstein, E.; Hansen, L.A.; Mallory, M.; Trojanowski, J.Q.; Galasko, D.; Masliah, E. Altered expression of the synuclein family mRNA in Lewy body and Alzheimer’s disease. Brain Res 2001, 914, 48–56. [Google Scholar]
- Beyer, K.; Domingo-Sabat, M.; Santos, C.; Tolosa, E.; Ferrer, I.; Ariza, A. The decrease of β-synuclein in cortical brain areas defines a molecular subgroup of dementia with Lewy bodies. Brain 2010, 133, 3724–3733. [Google Scholar]
- Brighina, L.; Okubadejo, N.U.; Schneider, N.K.; Lesnick, T.G.; de Andrade, M.; Cunningham, J.M.; Farrer, M.J.; Lincoln, S.J.; Rocca, W.A.; Maraganore, D.M. Beta-synuclein gene variants and Parkinson’s disease: A preliminary case-control study. Neurosci. Lett 2007, 420, 229–234. [Google Scholar]
- Nishioka, K.; Wider, C.; Vilarino-Guell, C.; Soto-Ortolaza, A.I.; Lincoln, S.J.; Kachergus, J.M.; Jasinska-Myga, B.; Ross, O.A.; Rajput, A.; Robinson, C.A.; et al. Association of alpha-, beta-, and gamma-Synuclein with diffuse lewy body disease. Arch. Neurol 2010, 67, 970–975. [Google Scholar]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar]
- Galvin, J.E.; Giasson, B.; Hurtig, H.I.; Lee, V.M.; Trojanowski, J.Q. Neurodegeneration with brain iron accumulation, type 1 is characterized by alpha-, beta-, and gamma-synuclein neuropathology. Am. J. Pathol 2000, 157, 361–368. [Google Scholar]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell. Biol 2002, 4, 160–164. [Google Scholar]
- Ellis, C.E.; Schwartzberg, P.L.; Grider, T.L.; Fink, D.W.; Nussbaum, R.L. Alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J. Biol. Chem 2001, 276, 3879–3884. [Google Scholar]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Invest 2009, 119, 3257–3265. [Google Scholar]
- Cole, R.N.; Hart, G.W. Cytosolic O-glycosylation is abundant in nerve terminals. J. Neurochem 2001, 79, 1080–1089. [Google Scholar]
- Bertoncini, C.W.; Rasia, R.M.; Lamberto, G.R.; Binolfi, A.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Structural characterization of the intrinsically unfolded protein beta-synuclein, a natural negative regulator of alpha-synuclein aggregation. J. Mol. Biol 2007, 372, 708–722. [Google Scholar]
- Aghazadeh, B.; Rosen, M.K. Ligand recognition by SH3 and WW domains: The role of N-alkylation in PPII helices. Chem. Biol 1999, 6, R241–R246. [Google Scholar]
- Yamin, G.; Munishkina, L.A.; Karymov, M.A.; Lyubchenko, Y.L.; Uversky, V.N.; Fink, A.L. Forcing nonamyloidogenic beta-synuclein to fibrillate. Biochemistry 2005, 44, 9096–9107. [Google Scholar]
- Beyer, K. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol 2006, 112, 237–251. [Google Scholar]
- Beyer, K.; Domingo-Sabat, M.; Lao, J.I.; Carrato, C.; Ferrer, I.; Ariza, A. Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics 2008, 9, 15–23. [Google Scholar]
- Beyer, K.; Munoz-Marmol, A.M.; Sanz, C.; Marginet-Flinch, R.; Ferrer, I.; Ariza, A. New brain-specific beta-synuclein isoforms show expression ratio changes in Lewy body diseases. Neurogenetics 2012, 13, 61–72. [Google Scholar]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Crews, L.; Bar-On, P.; Gage, F.H.; Marr, R.; Masliah, E. An antiaggregation gene therapy strategy for Lewy body disease utilizing beta-synuclein lentivirus in a transgenic model. Gene Ther 2004, 11, 1713–1723. [Google Scholar]
- Shaltiel-Karyo, R.; Frenkel-Pinter, M.; Egoz-Matia, N.; Frydman-Marom, A.; Shalev, D.E.; Segal, D.; Gazit, E. Inhibiting α-synuclein oligomerization by stable cell-penetrating β-synuclein fragments recovers phenotype of Parkinson’s disease model flies. PLoS One 2010, 5, e13863. [Google Scholar]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. [Google Scholar]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fujita, M.; Sekigawa, A.; Sekiyama, K.; Takamatsu, Y.; Hashimoto, M. Possible Alterations in β-Synuclein, the Non-Amyloidogenic Homologue of α-Synuclein, during Progression of Sporadic α-Synucleinopathies. Int. J. Mol. Sci. 2012, 13, 11584-11592. https://doi.org/10.3390/ijms130911584
Fujita M, Sekigawa A, Sekiyama K, Takamatsu Y, Hashimoto M. Possible Alterations in β-Synuclein, the Non-Amyloidogenic Homologue of α-Synuclein, during Progression of Sporadic α-Synucleinopathies. International Journal of Molecular Sciences. 2012; 13(9):11584-11592. https://doi.org/10.3390/ijms130911584
Chicago/Turabian StyleFujita, Masayo, Akio Sekigawa, Kazunari Sekiyama, Yoshiki Takamatsu, and Makoto Hashimoto. 2012. "Possible Alterations in β-Synuclein, the Non-Amyloidogenic Homologue of α-Synuclein, during Progression of Sporadic α-Synucleinopathies" International Journal of Molecular Sciences 13, no. 9: 11584-11592. https://doi.org/10.3390/ijms130911584
APA StyleFujita, M., Sekigawa, A., Sekiyama, K., Takamatsu, Y., & Hashimoto, M. (2012). Possible Alterations in β-Synuclein, the Non-Amyloidogenic Homologue of α-Synuclein, during Progression of Sporadic α-Synucleinopathies. International Journal of Molecular Sciences, 13(9), 11584-11592. https://doi.org/10.3390/ijms130911584