Common Fragile Sites: Genomic Hotspots of DNA Damage and Carcinogenesis


Abstract
:1. Introduction
2. Mechanisms of CFS Instability
3. Cancer-Associated CFS Genes
4. CFS Expression and Defects in Checkpoint Proteins
5. An Active Role of CACGs in Maintaining Genomic Stability
6. Clinical Significance of CACGs
7. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Schwartz, M.; Zlotorynski, E.; Kerem, B. The molecular basis of common and rare fragile sites. Cancer Lett 2006, 232, 13–26. [Google Scholar]
- Sutherland, G.R.; Baker, E.; Richards, R.I. Fragile sites still breaking. Trends Genet 1998, 14, 501–506. [Google Scholar]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet 1984, 67, 136–142. [Google Scholar]
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar]
- Yunis, J.J.; Soreng, A.L. Constitutive fragile sites and cancer. Science 1984, 226, 1199–1204. [Google Scholar]
- Denison, S.R.; Simper, R.K.; Greenbaum, I.F. How common are common fragile sites in humans: Interindividual variation in the distribution of aphidicolin-induced fragile sites. Cytogenet. Genome Res 2003, 101, 8–16. [Google Scholar]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet 2007, 41, 169–192. [Google Scholar]
- Hashash, N.; Johnson, A.L.; Cha, R.S. Regulation of fragile sites expression in budding yeast by MEC1, RRM3 and hydroxyurea. J. Cell Sci 2011, 124, 181–185. [Google Scholar]
- Glover, T.W.; Stein, C.K. Induction of sister chromatid exchanges at common fragile sites. Am. J. Hum. Genet 1987, 41, 882–890. [Google Scholar]
- Glover, T.W.; Stein, C.K. Chromosome breakage and recombination at fragile sites. Am. J. Hum. Genet 1988, 43, 265–273. [Google Scholar]
- Wang, N.D.; Testa, J.R.; Smith, D.I. Determination of the specificity of aphidicolin-induced breakage of the human 3p14.2 fragile site. Genomics 1993, 17, 341–347. [Google Scholar]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell. Biol 2009, 11, 753–760. [Google Scholar]
- Coquelle, A.; Pipiras, E.; Toledo, F.; Buttin, G.; Debatisse, M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell 1997, 89, 215–225. [Google Scholar]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar]
- Bartova, E.; Galiova, G.; Legartova, S.; Stixova, L.; Jugova, A.; Kozubek, S. Genomic instability in the context of chromatin structure and fragile sites. Crit. Rev. Eukaryot. Gene Expr 2010, 20, 181–194. [Google Scholar]
- Wilke, C.M.; Hall, B.K.; Hoge, A.; Paradee, W.; Smith, D.I.; Glover, T.W. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: Direct evidence for the coincidence of viral integration sites and fragile sites. Hum. Mol. Genet 1996, 5, 187–195. [Google Scholar]
- De Braekeleer, M.; Sreekantaiah, C.; Haas, O. Herpes simplex virus and human papillomavirus sites correlate with chromosomal breakpoints in human cervical carcinoma. Cancer Genet. Cytogenet 1992, 59, 135–137. [Google Scholar]
- Popescu, N.C.; DiPaolo, J.A. Preferential sites for viral integration on mammalian genome. Cancer Genet. Cytogenet 1989, 42, 157–137. [Google Scholar]
- Smith, P.P.; Friedman, C.; Bryant, E.M.; McDougall, J.K. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer 1992, 5, 150–157. [Google Scholar]
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R.; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet 1998, 7, 755–761. [Google Scholar]
- Wang, L.; Darling, J.; Zhang, J.S.; Huang, H.; Liu, W.; Smith, D.I. Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum. Mol. Genet 1999, 8, 431–437. [Google Scholar]
- Palakodeti, A.; Han, Y.; Jiang, Y.; Le Beau, M.M. The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer 2004, 39, 71–76. [Google Scholar]
- Hellman, A.; Rahat, A.; Scherer, S.W.; Darvasi, A.; Tsui, L.C.; Kerem, B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell. Biol 2000, 20, 4420–4427. [Google Scholar]
- Pelliccia, F.; Bosco, N.; Curatolo, A.; Rocchi, A. Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet. Genome Res 2008, 121, 196–200. [Google Scholar]
- Palumbo, E.; Matricardi, L.; Tosoni, E.; Bensimon, A.; Russo, A. Replication dynamics at common fragile site FAR6E. Chromosoma 2010, 119, 575–587. [Google Scholar]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol 2003, 23, 7143–7151. [Google Scholar]
- Mishmar, D.; Rahat, A.; Scherer, S.W.; Nyakatura, G.; Hinzmann, B.; Kohwi, Y.; Mandel-Gutfroind, Y.; Lee, J.R.; Drescher, B.; Sas, D.E.; et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. USA 1998, 95, 8141–8146. [Google Scholar]
- Tsai, A.G.; Engelhart, A.E.; Hatmal, M.M.; Houston, S.I.; Hud, N.V.; Haworth, I.S.; Lieber, M.R. Conformational variants of duplex DNA correlated with cytosine-rich chromosomal fragile sites. J. Biol. Chem 2009, 284, 7157–7164. [Google Scholar]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar]
- Pearson, C.E.; Edamura, K.N.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet 2005, 6, 729–742. [Google Scholar]
- Bichara, M.; Wagner, J.; Lambert, I.B. Mechanisms of tandem repeat instability in bacteria. Mutation Res 2006, 598, 144–163. [Google Scholar]
- Kang, S.; Jaworski, A.; Ohshima, K.; Wells, R.D. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat. Genet 1995, 10, 213–218. [Google Scholar]
- Ragland, R.L.; Glynn, M.W.; Arlt, M.F.; Glover, T.W. Stably transfected common fragile site sequences exhibit instability at ectopic sites. Genes Chromosomes Cancer 2008, 47, 860–872. [Google Scholar]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar]
- Palakodeti, A.; Lucas, I.; Jiang, Y.; Young, D.J.; Fernald, A.A.; Karrison, T.; Le Beau, M.M. Impaired replication dynamics at the FRA3B common fragile site. Hum. Mol. Genet 2010, 19, 99–110. [Google Scholar]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar]
- Ozeri-Galai, E.; Bester, A.C.; Kerem, B. The complex basis underlying common fragile site instability in cancer. Trends Genet 2012, 28, 295–302. [Google Scholar]
- Franchitto, A.; Pichierri, P. Understanding the molecular basis of common fragile sites instability: Role of the proteins involved in the recovery of stalled replication forks. Cell Cycle 2011, 10, 4039–4046. [Google Scholar]
- Le Tallec, B.; Dutrillaux, B.; Lachages, A.M.; Millot, G.A.; Brison, O.; Debatisse, M. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol 2011, 18, 1421–1423. [Google Scholar]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet 2012, 28, 22–32. [Google Scholar]
- Yunis, J.J; Soreng, A.L.; Bowe, A.E. Fragile sites are targets of diverse mutagens and carcinogens. Oncogene 1987, 1, 59–69. [Google Scholar]
- Druck, T.; Hadaczek, P.; Fu, T.B.; Ohta, M.; Siprashvili, Z.; Baffa, R.; Negrini, M.; Kastury, K.; Veronese, M.L.; Rosen, D.; et al. Structure and expression of the human FHIT gene in normal and tumor cells. Cancer Res 1997, 57, 504–512. [Google Scholar]
- Michael, D.; Beer, D.G.; Wilke, C.W.; Miller, D.E.; Glover, T.W. Frequent deletions of FHIT and FRA3B in Barrett’s metaplasia and esophageal adenocarcinomas. Oncogene 1997, 15, 1653–1659. [Google Scholar]
- Mimori, K.; Druck, T.; Inoue, H.; Alder, H.; Berk, L.; Mori, M.; Huebner, K.; Croce, C.M. Cancer-specific chromosome alterations in the constitutive fragile region FRA3B. Proc. Natl. Acad. Sci. USA 1999, 96, 7456–7461. [Google Scholar]
- Ohta, M.; Inoue, H.; Cotticelli, M.G.; Kastury, K.; Baffa, R.; Palazzo, J.; Siprashvili, Z.; Mori, M.; McCue, P.; Druck, T.; et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996, 84, 587–597. [Google Scholar]
- Roy, D.; Sin, S.H.; Damania, B.; Dittmer, D.P. Tumor suppressor genes FHIT and WWOX are deleted in primary effusion lymphoma (PEL) cell lines. Blood 2011, 118, e32–39. [Google Scholar]
- Huiping, C.; Kristjansdottir, S.; Bergthorsson, J.T.; Jonasson, J.G.; Magnusson, J.; Egilsson, V.; Ingvarsson, S. High frequency of LOH, MSI and abnormal expression of FHIT in gastric cancer. Eur. J. Cancer 2002, 38, 728–735. [Google Scholar]
- Barnes, L.D.; Garrison, P.N.; Siprashvili, Z.; Guranowski, A.; Robinson, A.K.; Ingram, S.W.; Croce, C.M.; Ohta, M.; Huebner, K. FHIT, a putative tumor suppressor in humans, is a dinucleoside 5′,5″′-P1,P3-triphosphate hydrolase. Biochemistry 1996, 35, 11529–11535. [Google Scholar]
- Zanesi, N.; Fidanza, V.; Fong, L.Y.; Mancini, R.; Druck, T.; Valtieri, M.; Rüdiger, T.; McCue, P.A.; Croce, C.M.; Huebner, K. The tumor spectrum in FHIT-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10250–10255. [Google Scholar]
- Siprashvili, Z.; Sozzi, G.; Barnes, L.D.; McCue, P.; Robinson, A.K.; Eryomin, V.; Sard, L.; Tagliabue, E.; Greco, A.; Fusetti, L.; et al. Replacement of FHIT in cancer cells suppresses tumorigenicity. Proc. Natl. Acad. Sci. USA 1997, 94, 13771–13776. [Google Scholar]
- Weiske, J.; Albring, K.F.; Huber, O. The tumor suppressor FHIT acts as a repressor of β-catenin transcriptional activity. Proc. Natl. Acad. Sci. USA 2007, 104, 20344–20349. [Google Scholar]
- Jayachandran, G.; Sazaki, J.; Nishizaki, M.; Xu, K.; Girard, L.; Minna, J.D.; Roth, J.A.; Ji, L. Fragile histidine triad-mediated tumor suppression of lung cancer by targeting multiple components of the Ras/Rho GTPase molecular switch. Cancer Res 2007, 67, 10379–10388. [Google Scholar]
- Bednarek, A.K.; Laflin, K.J.; Daniel, R.L.; Liao, Q.; Hawkins, K.A.; Aldaz, C.M. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3–24.1, a region frequently affected in breast cancer. Cancer Res 2000, 60, 2140–2145. [Google Scholar]
- Ried, K.; Finnis, M.; Hobson, L.; Mangelsdorf, M.; Dayan, S.; Nancarrow, J.K.; Woollatt, E.; Kremmidiotis, G.; Gardner, A.; Venter, D.; et al. Common chromosomal fragile site FRA16D sequence: Identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet 2000, 9, 1651–1663. [Google Scholar]
- Kuroki, T.; Trapasso, F.; Shiraishi, T.; Alder, H.; Mimori, K.; Mori, M.; Croce, C.M. Genetic alterations of the tumor suppressor gene WWOX in esophageal squamous cell carcinoma. Cancer Res 2002, 62, 2258–2260. [Google Scholar]
- Kuroki, T.; Yendamuri, S.; Trapasso, F.; Matsuyama, A.; Aqeilan, R.I.; Alder, H.; Rattan, S.; Cesari, R.; Nolli, M.L.; Williams, N.N.; et al. The tumor suppressor gene WWOX at FRA16D is involved in pancreatic carcinogenesis. Clin. Cancer Res 2004, 10, 2459–2465. [Google Scholar]
- Aqeilan, R.I.; Kuroki, T.; Pekarsky, Y.; Albagha, O.; Trapasso, F.; Baffa, R.; Huebner, K.; Edmonds, P.; Croce, C.M. Loss of WWOX expression in gastric carcinoma. Clin. Cancer Res 2004, 10, 3053–3058. [Google Scholar]
- Yendamuri, S.; Kuroki, T.; Trapasso, F.; Henry, A.C.; Dumon, K.R.; Huebner, K.; Williams, N.N.; Kaiser, L.R.; Croce, C.M. WW domain containing oxidoreductase gene expression is altered in non-small cell lung cancer. Cancer Res 2003, 63, 878–881. [Google Scholar]
- Watanabe, A.; Hippo, Y.; Taniguchi, H.; Iwanari, H.; Yashiro, M.; Hirakawa, K.; Kodama, T.; Aburatani, H. An opposing view on WWOX protein function as a tumor suppressor. Cancer Res 2003, 63, 8629–8633. [Google Scholar]
- Aqeilan, R.I.; Hagan, J.P.; Aqeilan, H.A.; Pichiorri, F.; Fong, L.Y.; Croce, C.M. Inactivation of the WWOX gene accelerates forestomach tumor progression in vivo. Cancer Res 2007, 67, 5606–5610. [Google Scholar]
- Aqeilan, R.I.; Trapasso, F.; Hussain, S.; Costinean, S.; Marshall, D.; Pekarsky, Y.; Hagan, J.P.; Zanesi, N.; Kaou, M.; Stein, G.S.; et al. Targeted deletion of WWOX reveals a tumor suppressor function. Proc. Natl. Acad. Sci. USA 2007, 104, 3949–3954. [Google Scholar]
- Gourley, C.; Paige, A.J.; Taylor, K.J.; Scott, D.; Francis, N.J.; Rush, R.; Aldaz, C.M.; Smyth, J.F.; Gabra, H. WWOX mRNA expression profile in epithelial ovarian cancer supports the role of WWOX variant 1 as a tumor suppressor, although the role of variant 4 remains unclear. Int. J. Oncol 2005, 26, 1681–1689. [Google Scholar]
- Bednarek, A.K.; Keck-Waggoner, C.L.; Daniel, R.L.; Laflin, K.J.; Bergsagel, P.L.; Kiguchi, K.; Brenner, A.J.; Aldaz, C.M. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Res 2001, 61, 8068–8073. [Google Scholar]
- Ludes-Meyers, J.H.; Bednarek, A.K.; Popescu, N.C.; Bedford, M.; Aldaz, C.M. WWOX, the common chromosomal fragile site, FRA16D, cancer gene. Cytogenet. Genome Res 2003, 100, 101–110. [Google Scholar]
- Chang, N.S.; Doherty, J.; Ensign, A.; Lewis, J.; Heath, J.; Schultz, L.; Chen, S.T.; Oppermann, U. Molecular mechanisms underlying WOX1 activation during apoptotic and stress responses. Biochem. Pharmacol 2003, 66, 1347–1354. [Google Scholar]
- Gaudio, E.; Palamarchuk, A.; Palumbo, T.; Trapasso, F.; Pekarsky, Y.; Croce, C.M.; Aqeilan, R.I. Physical association with WWOX suppresses c-Jun transcriptional activity. Cancer Res 2006, 66, 11585–11589. [Google Scholar]
- Limongi, M.Z.; Pelliccia, F.; Rocchi, A. Characterization of the human common fragile site FRA2G. Genomics 2003, 81, 93–97. [Google Scholar]
- Brueckner, L.M.; Sagulenko, E.; Hess, E.M.; Zheglo, D.; Blumrich, A.; Schwab, M.; Savelyeva, L. Genomic rearrangements at the FRA2H common fragile site frequently involve non-homologous recombination events across LTR and L1(LINE) repeats. Hum. Genet 2012, 131, 1345–1359. [Google Scholar]
- Rozier, L.; El-Achkar, E.; Apiou, F.; Debatisse, M. Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: Possible association with an inherited disease and cancer. Oncogene 2004, 23, 6872–6880. [Google Scholar]
- Denison, S.R.; Callahan, G.; Becker, N.A.; Phillips, L.A.; Smith, D.I. Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes Chromosomes Cancer 2003, 38, 40–52. [Google Scholar]
- Morelli, C.; Karayianni, E.; Magnanini, C.; Mungall, A.J.; Thorland, E.; Negrini, M.; Smith, D.I.; Barbanti-Brodano, G. Cloning and characterization of the common fragile site FRA6F harboring a replicative senescence gene and frequently deleted in human tumors. Oncogene 2002, 21, 7266–7276. [Google Scholar]
- Bosco, N.; Pelliccia, F.; Rocchi, A. Characterization of FRA7B, a human common fragile site mapped at the 7p chromosome terminal region. Cancer Genet. Cytogenet 2010, 202, 47–52. [Google Scholar]
- Huang, H.; Qian, C.; Jenkins, R.B.; Smith, D.I. Fish mapping of YAC clones at human chromosomal band 7q31.2: Identification of YACS spanning FRA7G within the common region of LOH in breast and prostate cancer. Genes Chromosomes Cancer 1998, 21, 152–159. [Google Scholar]
- Huang, H.; Reed, C.P.; Mordi, A.; Lomberk, G.; Wang, L.; Shridhar, V.; Hartmann, L.; Jenkins, R.; Smith, D.I. Frequent deletions within FRA7G at 7q31.2 in invasive epithelial ovarian cancer. Genes Chromosomes Cancer 1999, 24, 48–55. [Google Scholar]
- Miller, C.T.; Lin, L.; Casper, A.M.; Lim, J.; Thomas, D.G.; Orringer, M.B.; Chang, A.C.; Chambers, A.F.; Giordano, T.J.; Glover, T.W.; et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006, 25, 409–418. [Google Scholar]
- Ciullo, M.; Debily, M.A.; Rozier, L.; Autiero, M.; Billault, A.; Mayau, V.; El Marhomy, S.; Guardiola, J.; Bernheim, A.; Coullin, P.; et al. Initiation of the breakage-fusion-bridge mechanism through common fragile site activation in human breast cancer cells: The model of PIP gene duplication from a break at FRA7I. Hum. Mol. Genet 2002, 11, 2887–2894. [Google Scholar]
- Helmrich, A.; Stout-Weider, K.; Matthaei, A.; Hermann, K.; Heiden, T.; Schrock, E. Identification of the human/mouse syntenic common fragile site FRA7K/Fra12C1: Relation of FRA7K and other human common fragile sites on chromosome 7 to evolutionary breakpoints. Int. J. Cancer 2007, 120, 48–54. [Google Scholar]
- Ferber, M.J.; Eilers, P.; Schuuring, E.; Fenton, J.A.; Fleuren, G.J.; Kenter, G.; Szuhai, K.; Smith, D.I.; Raap, A.K.; Brink, A.A. Positioning of cervical carcinoma and Burkitt lymphoma translocation breakpoints with respect to the human papillomavirus integration cluster in FRA8C at 8q24.13. Cancer Genet. Cytogenet 2004, 154, 1–9. [Google Scholar]
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242. [Google Scholar]
- Callahan, G.; Denison, S.R.; Phillips, L.A.; Shridhar, V.; Smith, D.I. Characterization of the common fragile site FRA9E and its potential role in ovarian cancer. Oncogene 2003, 22, 590–601. [Google Scholar]
- Li, Z.; Zhang, Q.; Mao, J.H.; Weise, A.; Mrasek, K.; Fan, X.; Zhang, X.; Liehr, T.; Lu, K.H.; Balmain, A.; et al. An HDAC1-binding domain within FATS bridges p21 turnover to radiation-induced tumorigenesis. Oncogene 2010, 29, 2659–2671. [Google Scholar]
- Gandhi, M.; Dillon, L.W.; Pramanik, S.; Nikiforov, Y.E.; Wang, Y.H. DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene 2010, 29, 2272–2280. [Google Scholar]
- Gandhi, M.; Medvedovic, M.; Stringer, J.R.; Nikiforov, Y.E. Interphase chromosome folding determines spatial proximity of genes participating in carcinogenic RET/PTC rearrangements. Oncogene 2006, 25, 2360–2366. [Google Scholar]
- Arlt, M.F.; Miller, D.E.; Beer, D.G.; Glover, T.W. Molecular characterization of FRAXB and comparative common fragile site instability in cancer cells. Genes Chromosomes Cancer 2002, 33, 82–92. [Google Scholar]
- McAvoy, S.; Ganapathiraju, S.; Perez, D.S.; James, C.D.; Smith, D.I. DMD and IL1RAPL1: Two large adjacent genes localized within a common fragile site (FRAXC) have reduced expression in cultured brain tumors. Cytogenet. Genome Res 2007, 119, 196–203. [Google Scholar]
- Mitsui, J.; Takahashi, Y.; Goto, J.; Tomiyama, H.; Ishikawa, S.; Yoshino, H.; Minami, N.; Smith, D.I.; Lesage, S.; Aburatani, H.; et al. Mechanisms of genomic instabilities underlying two common fragile-site-associated loci, PARK2 and DMD, in germ cell and cancer cell lines. Am. J. Hum. Genet 2010, 87, 75–89. [Google Scholar]
- Nam, H.J.; Chae, S.; Jang, S.H.; Cho, H.; Lee, J.H. The PI3K-Akt mediates oncogenic Met-induced centrosome amplification and chromosome instability. Carcinogenesis 2010, 31, 1531–1540. [Google Scholar]
- Joo, K.M.; Jin, J.; Kim, E.; Ho Kim, K.; Kim, Y.; Gu Kang, B.; Kang, Y.J.; Lathia, J.D.; Ho Cheong, K.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res 2012, 72, 3828–3838. [Google Scholar]
- Tatarelli, C.; Linnenbach, A.; Mimori, K.; Croce, C.M. Characterization of the human TESTIN gene localized in the FRA7G region at 7q31.2. Genomics 2000, 68, 1–12. [Google Scholar]
- Sarti, M.; Sevignani, C.; Calin, G.A.; Aqeilan, R.; Shimizu, M.; Pentimalli, F.; Picchio, M.C.; Godwin, A.; Rosenberg, A.; Drusco, A.; et al. Adenoviral transduction of TESTIN gene into breast and uterine cancer cell lines promotes apoptosis and tumor reduction in vivo. Clin. Cancer Res 2005, 11, 806–813. [Google Scholar]
- Han, S.Y.; Druck, T.; Huebner, K. Candidate tumor suppressor genes at FRA7G are coamplified with MET and do not suppress malignancy in a gastric cancer. Genomics 2003, 81, 105–107. [Google Scholar]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar]
- Nyegaard, M.; Overgaard, M.T.; Su, Y.Q.; Hamilton, A.E.; Kwintkiewicz, J.; Hsieh, M.; Nayak, N.R.; Conti, M.; Conover, C.A.; Giudice, L.C. Lack of functional pregnancy-associated plasma protein-A (PAPPA) compromises mouse ovarian steroidogenesis and female fertility. Biol. Reprod 2010, 82, 1129–1138. [Google Scholar]
- Santoro, M.; Melillo, R.M.; Fusco, A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology prize lecture. Eur. J. Endoc 2006, 155, 645–653. [Google Scholar]
- Dillon, L.W.; Lehman, C.E.; Wang, Y.H. The role of fragile sites in sporadic papillary thyroid carcinoma. J. Thyroid Res 2012, 2012, 927683. [Google Scholar]
- Dillon, L.W.; Burrow, A.A.; Wang, Y.H. DNA instability at chromosomal fragile sites in cancer. Curr. Genomics 2010, 11, 326–337. [Google Scholar]
- Luo, Y.; Tsuchiya, K.D., Il; Park, D.; Fausel, R.; Kanngurn, S.; Welcsh, P.; Dzieciatkowski, S.; Wang, J.; Grady, W.M. RET is a potential tumor suppressor gene in colorectal cancer. Oncogene 2012. [Google Scholar] [CrossRef]
- Wagner, S.M.; Zhu, S.; Nicolescu, A.C.; Mulligan, L.M. Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2. Clinics (Sao Paulo) 2012, 67, 77–84. [Google Scholar]
- Poulogiannis, G.; McIntyre, R.E.; Dimitriadi, M.; Apps, J.R.; Wilson, C.H.; Ichimura, K.; Luo, F.; Cantley, L.C.; Wyllie, A.H.; Adams, D.J.; et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15145–15150. [Google Scholar]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet 2010, 42, 77–82. [Google Scholar]
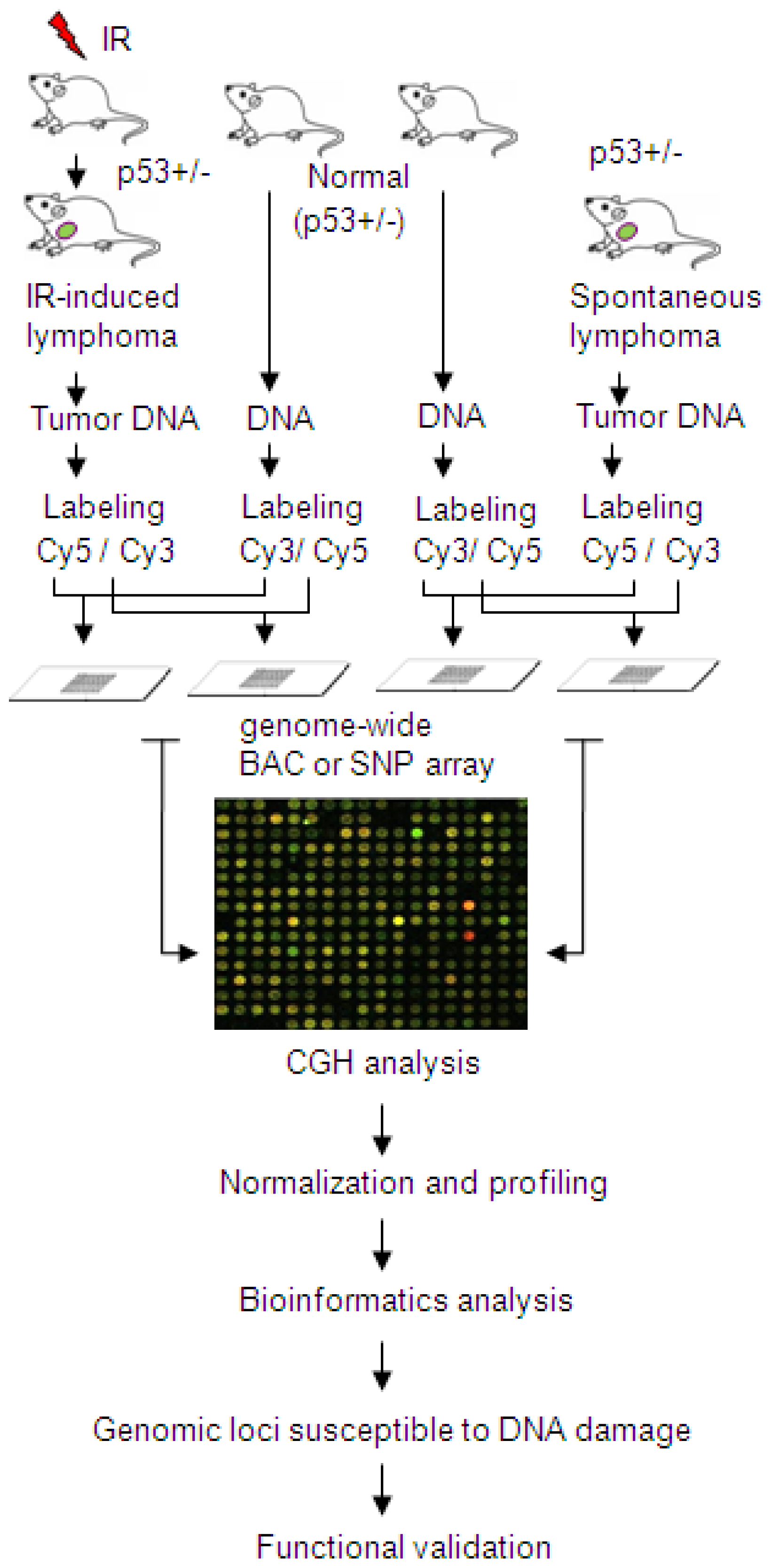
- Mao, J.H.; Li, J.; Jiang, T.; Li, Q.; Wu, D.; Perez-Losada, J.; DelRosario, R.; Peterson, L.; Balmain, A.; Cai, W.W. Genomic instability in radiation-induced mouse lymphoma from p53 heterozygous mice. Oncogene 2005, 24, 7924–7934. [Google Scholar]
- Kemp, C.J.; Wheldon, T.; Balmain, A. p53-Deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat. Genet 1994, 8, 66–69. [Google Scholar]
- Cai, W.W.; Mao, J.H.; Chow, C.W.; Damani, S.; Balmain, A.; Bradley, A. Genome-wide detection of chromosomal imbalances in tumors using BAC microarrays. Nat. Biotechnol 2002, 20, 393–396. [Google Scholar]
- Maier, D.; Comparone, D.; Taylor, E.; Zhang, Z.; Gratzl, O.; van Meir, E.G.; Scott, R.J.; Merlo, A. New deletion in low-grade oligodendroglioma at the glioblastoma suppressor locus on chromosome 10q25–26. Oncogene 1997, 15, 997–1000. [Google Scholar]
- Nagase, S.; Yamakawa, H.; Sato, S.; Yajima, A.; Horii, A. Identification of a 790-kilobase region of common allelic loss in chromosome 10q25-q26 in human endometrial cancer. Cancer Res 1997, 57, 1630–1633. [Google Scholar]
- Zhang, X.; Zhang, Q.; Zhang, J.; Qiu, L.; Yan, S.S.; Feng, J.; Sun, Y.; Huang, X.; Lu, K.H.; Li, Z. FATS is a transcriptional target of p53 and associated with antitumor activity. Mol. Cancer 2010, 9, 244. [Google Scholar]
- Zhang, J.; Gu, L.; Zhao, L.J.; Zhang, X.F.; Qiu, L.; Li, Z. Expression level of novel tumor suppressor gene FATS is associated with the outcome of node positive breast cancer. Chin. Med. J 2011, 124, 2894–2898. [Google Scholar]
- Tian, Y.; Zhang, J.; Yan, S.; Qiu, L.; Li, Z. FATS expression is associated with cisplatin sensitivity in non small cell lung cancer. Lung Cancer 2012, 76, 416–422. [Google Scholar]
- Mrasek, K.; Schoder, C.; Teichmann, A.C.; Behr, K.; Franze, B.; Wilhelm, K.; Blaurock, N.; Claussen, U.; Liehr, T.; Weise, A. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int. J. Oncol 2010, 36, 929–940. [Google Scholar]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions: A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar]
- Harrison, J.C.; Haber, J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet 2006, 40, 209–235. [Google Scholar]
- Glover, T.W.; Coyle-Morris, J.; Morgan, R. Fragile sites: Overview, occurrence in acute nonlymphocytic leukemia and effects of caffeine on expression. Cancer Genet. Cytogenet 1986, 19, 141–150. [Google Scholar]
- Lemoine, F.J.; Degtyareva, N.P.; Kokoska, R.J.; Petes, T.D. Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol. Cell. Biol 2008, 28, 5359–5368. [Google Scholar]
- Rey, L.; Sidorova, J.M.; Puget, N.; Boudsocq, F.; Biard, D.S.; Monnat, R.J., Jr; Cazaux, C.; Hoffmann, J.S. Human DNA polymerase eta is required for common fragile site stability during unperturbed DNA replication. Mol. Cell. Biol. 2009, 29, 3344–3354. [Google Scholar]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar]
- Ozeri-Galai, E.; Schwartz, M.; Rahat, A.; Kerem, B. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene 2008, 27, 2109–2117. [Google Scholar]
- Wan, C.; Kulkarni, A.; Wang, Y.H. ATR preferentially interacts with common fragile site FRA3B and the binding requires its kinase activity in response to aphidicolin treatment. Mutat. Res 2010, 686, 39–46. [Google Scholar]
- Durkin, S.G.; Arlt, M.F.; Howlett, N.G.; Glover, T.W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006, 25, 4381–4388. [Google Scholar]
- Zhu, M.; Weiss, R.S. Increased common fragile site expression, cell proliferation defects, and apoptosis following conditional inactivation of mouse Hus1 in primary cultured cells. Mol. Biol. Cell 2007, 18, 1044–1055. [Google Scholar]
- Musio, A.; Montagna, C.; Mariani, T.; Tilenni, M.; Focarelli, M.L.; Brait, L.; Indino, E.; Benedetti, P.A.; Chessa, L.; Albertini, A.; et al. SMC1 involvement in fragile site expression. Hum. Mol. Genet 2005, 14, 525–533. [Google Scholar]
- Arlt, M.F.; Xu, B.; Durkin, S.G.; Casper, A.M.; Kastan, M.B.; Glover, T.W. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Mol. Cell. Biol 2004, 24, 6701–6709. [Google Scholar]
- Focarelli, M.L.; Soza, S.; Mannini, L.; Paulis, M.; Montecucco, A.; Musio, A. Claspin inhibition leads to fragile site expression. Genes Chromosomes Cancer 2009, 48, 1083–1090. [Google Scholar]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet 2005, 14, 693–701. [Google Scholar]
- Schoder, C.; Liehr, T.; Velleuer, E.; Wilhelm, K.; Blaurock, N.; Weise, A.; Mrasek, K. New aspects on chromosomal instability: Chromosomal break-points in Fanconi anemia patients co-localize on the molecular level with fragile sites. Int. J. Oncol 2010, 36, 307–312. [Google Scholar]
- Cangi, M.G.; Piccinin, S.; Pecciarini, L.; Talarico, A.; Dal Cin, E.; Grassi, S.; Grizzo, A.; Maestro, R.; Doglioni, C. Constitutive overexpression of CDC25A in primary human mammary epithelial cells results in both defective DNA damage response and chromosomal breaks at fragile sites. Int. J. Cancer 2008, 123, 1466–1471. [Google Scholar]
- Vernole, P.; Muzi, A.; Volpi, A.; Terrinoni, A.; Dorio, A.S.; Tentori, L.; Shah, G.M.; Graziani, G. Common fragile sites in colon cancer cell lines: Role of mismatch repair, RAD51 and poly(ADP-ribose) polymerase-1. Mutat. Res 2011, 712, 40–48. [Google Scholar]
- Cheng, E.; Vaisica, J.A.; Ou, J.; Baryshnikova, A.; Lu, Y.; Roth, F.P.; Brown, G.W. Genome rearrangements caused by depletion of essential DNA replication proteins in Saccharomyces cerevisiae. Genetics 2012, 192, 147–160. [Google Scholar]
- Pirzio, L.M.; Pichierri, P.; Bignami, M.; Franchitto, A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol 2008, 180, 305–314. [Google Scholar]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol 2009, 11, 1315–1324. [Google Scholar]
- Arlt, M.F.; Glover, T.W. Inhibition of topoisomerase I prevents chromosome breakage at common fragile sites. DNA Repair 2010, 9, 678–689. [Google Scholar]
- Murfuni, I.; De Santis, A.; Federico, M.; Bignami, M.; Pichierri, P.; Franchitto, A. Perturbed replication induced genome-wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis 2012, 33, 1655–1663. [Google Scholar]
- Shah, S.N.; Opresko, P.L.; Meng, X.; Lee, M.Y.; Eckert, K.A. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic. Acids Res 2010, 38, 1149–1162. [Google Scholar]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov 2002, 1, 287–299. [Google Scholar]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res 2007, 5, 981–989. [Google Scholar]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995, 377, 552–557. [Google Scholar]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J. P.; Sedivy, J. M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995, 82, 675–684. [Google Scholar]
- Yan, S.; Ma, K.; Qiu, L.; Zhang, J.; Zhang, X.; Hao, X.; Li, Z. FATS is an E2-independent ubiquitin ligase that stabilizes p53 and promotes p53-dependent checkpoint response. To be submitted for publication.
- Thavathiru, E.; Ludes-Meyers, J.H.; MacLeod, M.C.; Aldaz, C.M. Expression of common chromosomal fragile site genes, WWOX/FRA16D and FHIT/FRA3B is downregulated by exposure to environmental carcinogens, UV, and BPDE but not by IR. Mol. Carcinog 2005, 44, 174–182. [Google Scholar]
- Durkin, S.G.; Ragland, R.L.; Arlt, M.F.; Mulle, J.G.; Warren, S.T.; Glover, T.W. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. USA 2008, 105, 246–251. [Google Scholar]
- Geradts, J.; Fong, K.M.; Zimmerman, P.V.; Minna, J.D. Loss of FHIT expression in non-small-cell lung cancer: Correlation with molecular genetic abnormalities and clinicopathological features. Br. J. Cancer 2000, 82, 1191–1197. [Google Scholar]
- Lee, Y.C.; Wu, C.T.; Shih, J.Y.; Jou, Y.S.; Chang, Y.L. Frequent allelic deletion at the FHIT locus associated with p53 overexpression in squamous cell carcinoma subtype of Taiwanese non-small-cell lung cancers. Br. J. Cancer 2004, 90, 2378–2383. [Google Scholar]
- Nunez, M.I.; Rosen, D.G.; Ludes-Meyers, J.H.; Abba, M.C.; Kil, H.; Page, R.; Klein-Szanto, A.J.; Godwin, A.K.; Liu, J.; Mills, G.B.; et al. WWOX protein expression varies among ovarian carcinoma histotypes and correlates with less favorable outcome. BMC Cancer 2005, 5, 64. [Google Scholar]
- Kaposi-Novak, P.; Lee, J.S.; Gòmez-Quiroz, L.; Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J. Clin. Invest 2006, 116, 1582–1595. [Google Scholar]
- Boice, J.D., Jr; Morin, M.M.; Glass, A.G.; Friedman, G.D.; Stovall, M.; Hoover, R.N.; Fraumeni, J.F., Jr. Diagnostic X-ray procedures and risk of leukemia, lymphoma, and multiple myeloma. JAMA 1991, 265, 1290–1294. [Google Scholar]
- Ron, E. Ionizing radiation and cancer risk: Evidence from epidemiology. Pediatr. Radiol 2002, 32, 232–237. [Google Scholar]
- Pearce, M.S.; Salotti, J.A.; Little, M.P.; McHugh, K.; Lee, C.; Kim, K.P.; Howe, N.L.; Ronckers, C.M.; Rajaraman, P.; Craft, A.W.; et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet 2012, 380, 499–505. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Human CFS | Location | Frequency | Associated genes | CACG |
|---|---|---|---|---|
| FRA2G | 2q31 | modest | IGRP, RDHL, LRP2 and others | not validated |
| FRA2H | 2q32 | modest | non-coding RNA gene | not validated |
| FRA3B | 3p14.2 | high | FHIT | FHIT |
| FRA4F | 4q22 | modest | GRID2 | not validated |
| FRA6E | 6q26 | modest | PARK2, PLG, LPA and others | PARK2 |
| FRA6F | 6q21 | Modest | REV3L, DIF13, FKHRL and others | not validated |
| FRA7B | 7p22 | low | THSD7A, SDK1, MAD1L1 | not validated |
| FRA7G | 7q31.2 | modest | MET, TESTIN, CAV, and others | MET, TESTIN |
| FRA7I | 7q36 | modest | PIP | not validated |
| FRA7K | 7q31 | modest | IMMP2L | not validated |
| FRA8C | 8q24 | modest | MYC | MYC |
| FRA9E | 9q32 | low | PAPPA and others | PAPPA |
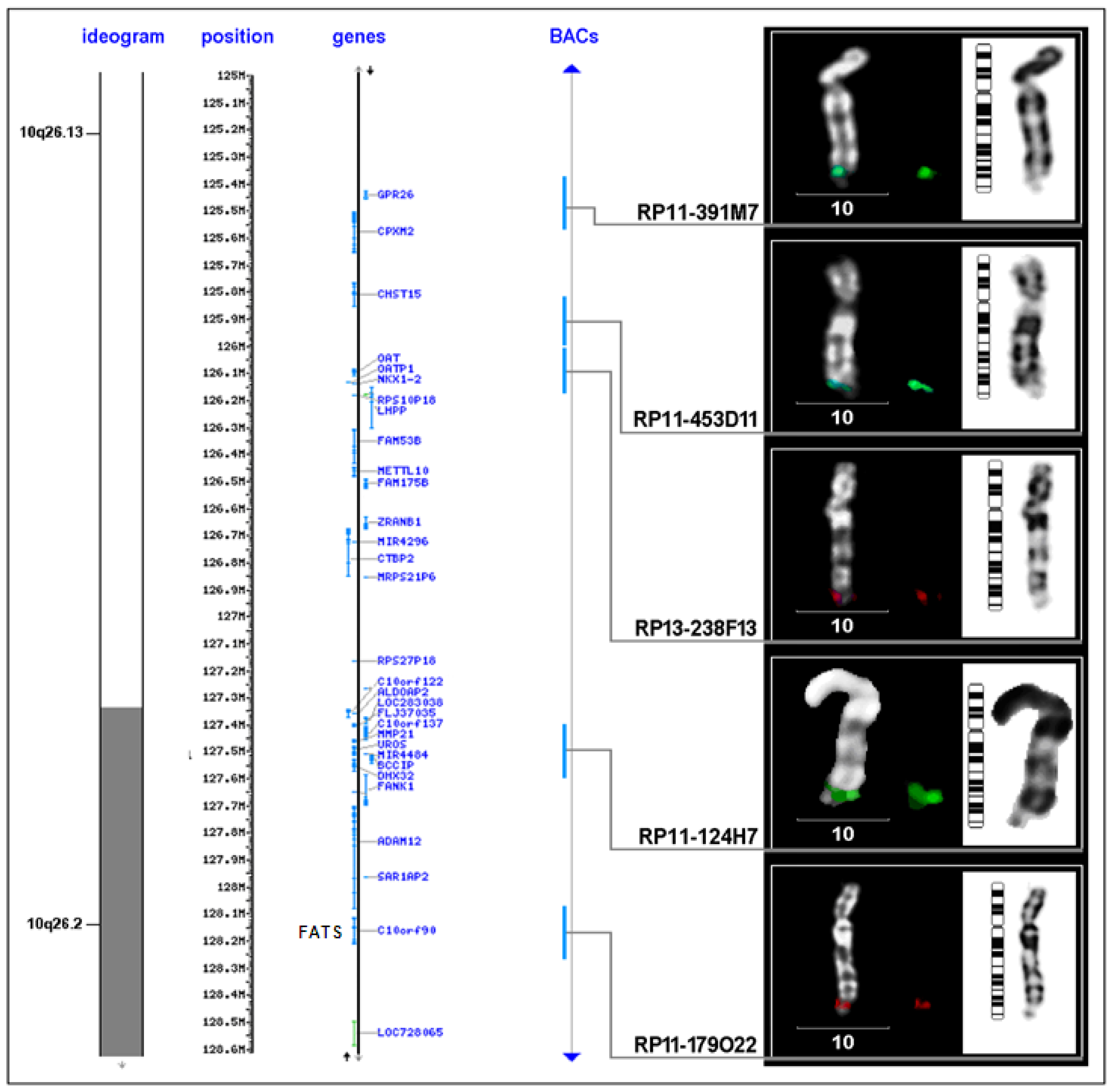
| FRA10F | 10q26 | low | FATS and others | FATS |
| FRA10G | 10q11 | low | RET, NCOA4 | RET |
| FRA16D | 16q23.2 | high | WWOX/FOR | WWOX |
| FRAXB | Xp22.3 | modest | STS, GS1 | not validated |
| FRAXC | Xq22.1 | modest | DMD, IL1RAPL1 | DMD |
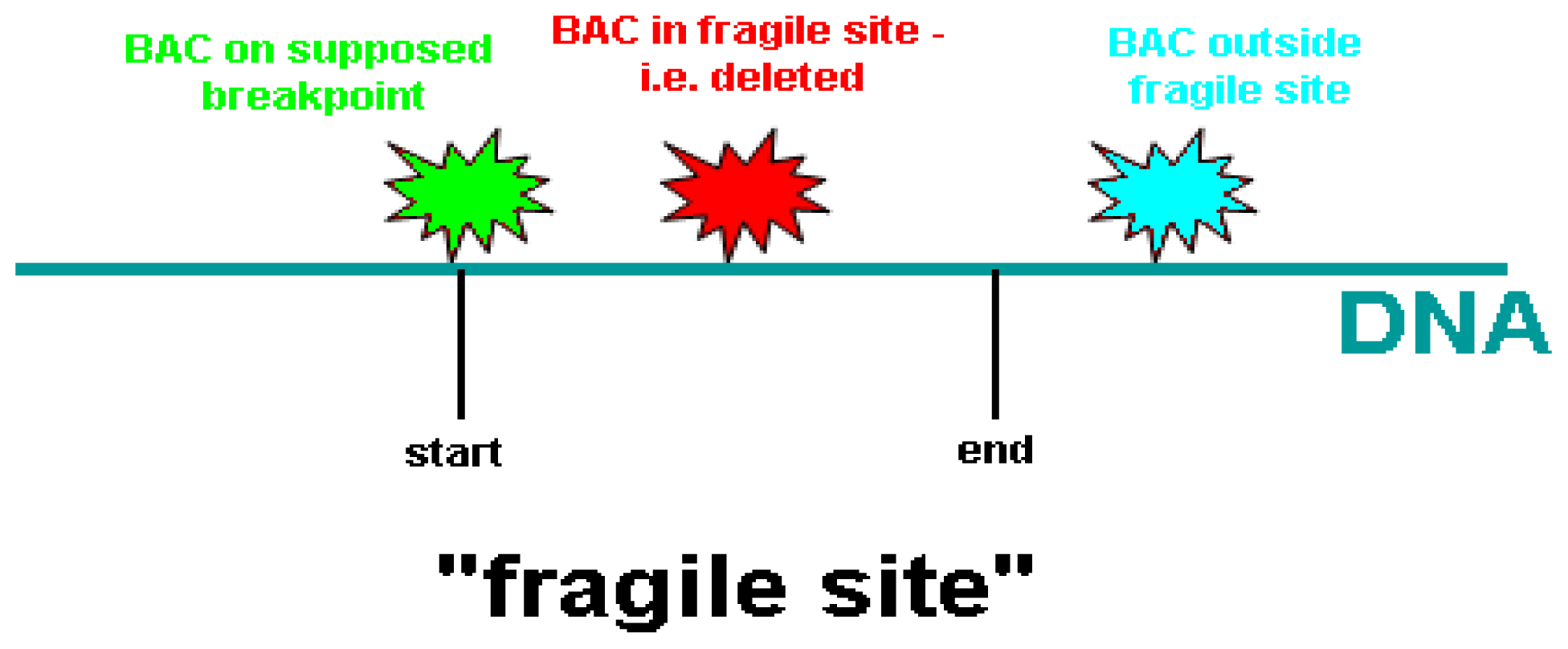
| BAC clone | Location on chromosome 10 (Mb) | Proximal to FRA10F | Supposed breakpoint | Inside FRA10F | Distal to FRA10F |
|---|---|---|---|---|---|
| RP11-198M6 | 121.577.828-121.765.053 | 1 | - | - | - |
| RP11-323P17 | 122.469.430-122.626.379 | 1 | - | - | - |
| RP11-105F10 | 123.764.471-123.939.147 | 1 | - | - | - |
| RP11-296H2 | 123.939.148-124.083.531 | 1 | - | - | - |
| RP11-436O19 | 124.267.798-124.268.644 | 1 | - | 1 | - |
| RP11-481L19 | 124.268.654-124.444.805 | 1 | - | - | - |
| RP11-564D11 | 124.592.597-124.787.891 | 1 | - | - | - |
| RP11-162A23 | 124.799.045-124.978.916 | 1 | - | - | - |
| RP11-391M7 | 125.381.493-125.576.300 | 1 | - | - | - |
| RP11-435D11 | 125.909.463-126.009.423 | - | 1 | - | - |
| RP13-238F13 | 126.009.424-126.190.394 | - | - | 1 | - |
| RP11-124H7 | 127.445.890-127.599.588 | - | 6 | - | - |
| RP11-179O22 | 128.076.478-128.246.854 | - | 1 | 3 | - |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, K.; Qiu, L.; Mrasek, K.; Zhang, J.; Liehr, T.; Quintana, L.G.; Li, Z. Common Fragile Sites: Genomic Hotspots of DNA Damage and Carcinogenesis. Int. J. Mol. Sci. 2012, 13, 11974-11999. https://doi.org/10.3390/ijms130911974
Ma K, Qiu L, Mrasek K, Zhang J, Liehr T, Quintana LG, Li Z. Common Fragile Sites: Genomic Hotspots of DNA Damage and Carcinogenesis. International Journal of Molecular Sciences. 2012; 13(9):11974-11999. https://doi.org/10.3390/ijms130911974
Chicago/Turabian StyleMa, Ke, Li Qiu, Kristin Mrasek, Jun Zhang, Thomas Liehr, Luciana Gonçalves Quintana, and Zheng Li. 2012. "Common Fragile Sites: Genomic Hotspots of DNA Damage and Carcinogenesis" International Journal of Molecular Sciences 13, no. 9: 11974-11999. https://doi.org/10.3390/ijms130911974
APA StyleMa, K., Qiu, L., Mrasek, K., Zhang, J., Liehr, T., Quintana, L. G., & Li, Z. (2012). Common Fragile Sites: Genomic Hotspots of DNA Damage and Carcinogenesis. International Journal of Molecular Sciences, 13(9), 11974-11999. https://doi.org/10.3390/ijms130911974