The Role of Crowded Physiological Environments in Prion and Prion-like Protein Aggregation

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Protein Aggregation in vitro
1.2. Macromolecular Crowding and Protein Aggregation in vitro
2. The Role of Crowded Physiological Environments in Prion Protein Aggregation
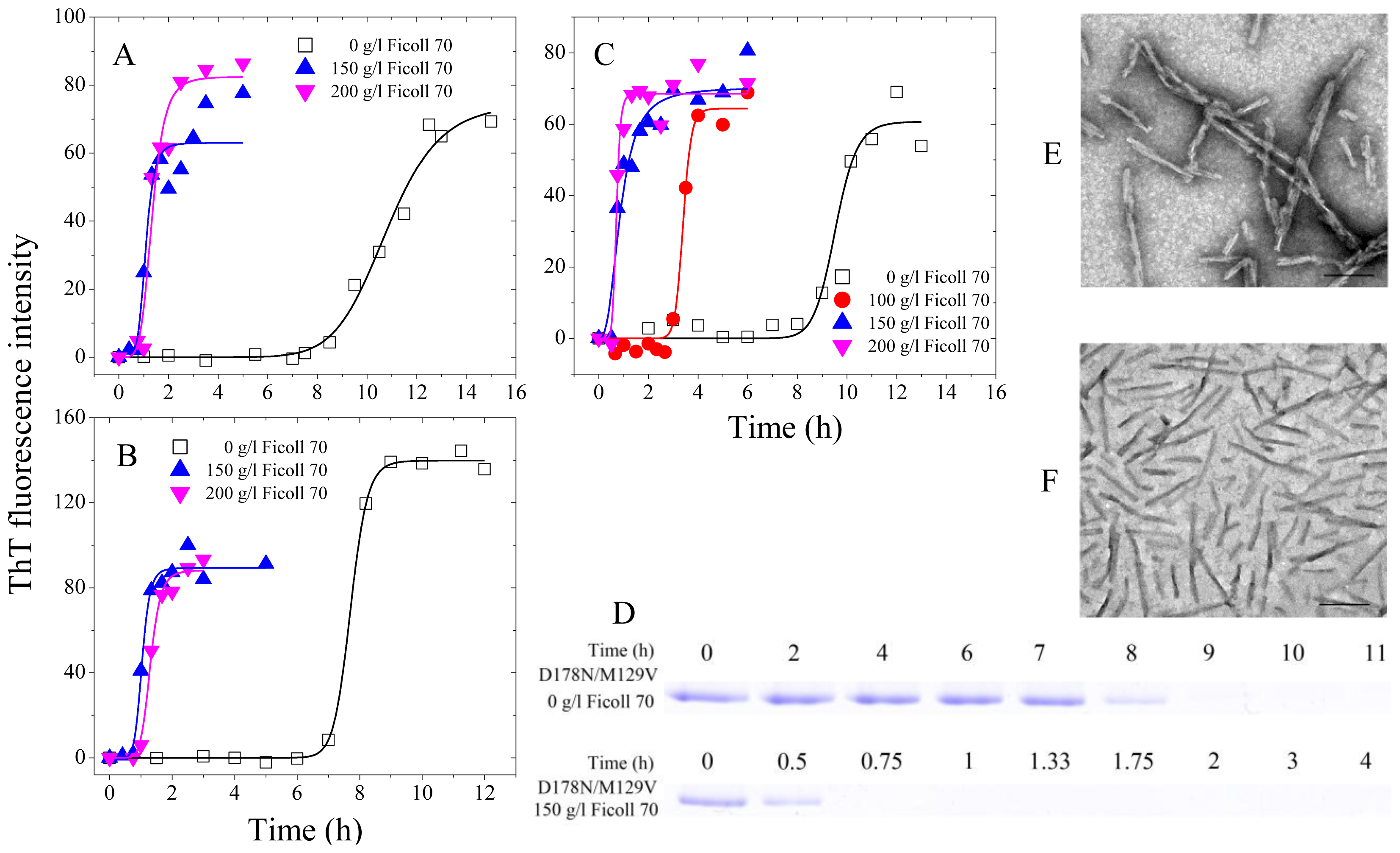
2.1. Human Prion Protein and Its Pathological Mutants
2.2. Rabbit Prion Protein
3. The Role of Crowded Physiological Environments in Prion-like Protein Aggregation
3.1. Tau Protein
3.2. α-Synuclein
3.3. Copper, Zinc Superoxide Dismutase (SOD1)
3.4. Amyloid-β
3.5. Other Proteins
4. Conclusions


Acknowledgments
Conflicts of Interest
References
- Zhou, H.X.; Rivas, G.; Minton, A.P. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys 2008, 37, 375–397. [Google Scholar]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci 2001, 26, 597–604. [Google Scholar]
- Bellotti, V.; Chiti, F. Amyloidogenesis in its biological environment: Challenging a fundamental issue in protein misfolding diseases. Curr. Opin. Struct. Biol 2008, 18, 771–779. [Google Scholar]
- Wickner, S.; Maurizi, M.R.; Gottesman, S. Posttranslational quality control: Folding, refolding, and degrading proteins. Science 1999, 286, 1888–1893. [Google Scholar]
- Lee, S.J.; Desplats, P.; Sigurdson, C.; Tsigelny, I.; Masliah, E. Cell-to-cell transmission of non-prion protein aggregates. Nat. Rev. Neurol 2010, 6, 702–706. [Google Scholar]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative diseases. Nat. Med 2004, S10–S17. [Google Scholar]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci 1993, 16, 460–465. [Google Scholar]
- Polymenidou, M.; Cleveland, D.W. The seeds of neurodegeneration: Prion- like spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar]
- Ellis, R.J.; Minton, A.P. Protein aggregation in crowded environments. Biol. Chem 2006, 387, 485–497. [Google Scholar]
- White, D.A.; Buell, A.K.; Knowles, T.P.; Welland, M.E.; Dobson, C.M. Protein aggregation in crowded environments. J. Am. Chem. Soc 2009, 132, 5170–5175. [Google Scholar]
- Zhou, Z.; Fan, J.B.; Zhu, H.L.; Shewmaker, F.; Yan, X.; Chen, X.; Chen, J.; Liang, Y. Crowded cell- like environment accelerates the nucleation step of amyloidogenic protein misfolding. J. Biol. Chem 2009, 284, 30148–30158. [Google Scholar]
- Ma, Q.; Fan, J.B.; Zhou, Z.; Zhou, B.R.; Meng, S.R.; Hu, J.Y.; Chen, J.; Liang, Y. The contrasting effect of macromolecular crowding on amyloid fibril formation. PloS One 2012, 7, e36288. [Google Scholar]
- Zhou, Z.; Yan, X.; Pan, K.; Chen, J.; Xie, Z.S.; Xiao, G.F.; Yang, F.Q.; Liang, Y. Fibril formation of the rabbit/human/bovine prion proteins. Biophys. J 2011, 101, 1483–1492. [Google Scholar]
- Sukenik, S.; Politi, R.; Ziserman, L.; Danino, D.; Friedler, A.; Harries, D. Crowding alone cannot account for cosolute effect on amyloid aggregation. PloS One 2011, 6, e15608. [Google Scholar]
- Lee, C.F.; Bird, S.; Shaw, M.; Jean, L.; Vaux, D.J. Combined effects of a gitation, macromolecular crowding, and interfaces on amyloidogenesis. J. Biol. Chem 2012, 287, 38006–38019. [Google Scholar]
- Cino, E.A.; Karttunen, M.; Choy, W.Y. Effects of molecular crowding on the dynamics of intrinsically disordered proteins. PloS One 2012, 7, e49876. [Google Scholar]
- Shtilerman, M.D.; Ding, T.T.; Lansbury, P.T., Jr. Molecular crowding accelerates fibrillization of α-synuclein: Could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry 2002, 41, 3855–3860. [Google Scholar]
- Uversky, V.N.; Cooper, E.M.; Bower, K.S.; Li, J.; Fink, A.L. Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett 2002, 515, 99–103. [Google Scholar]
- Munishkina, L.A.; Cooper, E.M.; Uversky, V.N.; Fink, A.L. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit 2004, 17, 456–464. [Google Scholar]
- Munishkina, L.A.; Ahmad, A.; Fink, A.L.; Uversky, V.N. Guiding protein aggregation with macromolecular crowding. Biochemistry 2008, 47, 8993–9006. [Google Scholar]
- Mo, Z.Y.; Zhu, Y.Z.; Zhu, H.L.; Fan, J.B.; Chen, J.; Liang, Y. Low micromolar zinc accelerates the fibrillization of human Tau via bridging of Cys-291 and Cys-322. J. Biol. Chem 2009, 284, 34648–34657. [Google Scholar]
- Zhu, H.L.; Fernández, C.; Fan, J.B.; Shewmaker, F.; Chen, J.; Minton, A.P.; Liang, Y. Quantitative characterization of heparin binding to Tau protein: Implication for inducer-mediated Tau filament formation. J. Biol. Chem 2010, 285, 3592–3299. [Google Scholar]
- Zhu, H.L.; Meng, S.R.; Fan, J.B.; Chen, J.; Liang, Y. Fibrillization of human Tau is accelerated by exposure to lead via interaction with His-330 and His-362. PloS One 2011, 6, e25020. [Google Scholar]
- Meng, S.R.; Zhu, Y.Z.; Guo, T.; Liu, X.L.; Chen, J.; Liang, Y. Fibril- forming motifs are essential and sufficient for the fibrillization of human Tau. PloS One 2012, 7, e38903. [Google Scholar]
- Pérez, M.; Valpuesta, J.M.; Medina, M.; Montejo de Garcini, E.; Avila, J. Polymerization of Tau into filaments in the presence of heparin: The minimal sequence required for Tau-Tau interaction. J. Neurochem 1996, 67, 1183–1190. [Google Scholar]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein Tau into Alzheimer- like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar]
- Friedhoff, P.; Schneider, A.; Mandelkow, E.M.; Mandelkow, E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein Tau monitored by fluorescence in solution. Biochemistry 1998, 37, 10223–10230. [Google Scholar]
- Paudel, H.K.; Li, W. Heparin- induced conformational change in microtubule-associated protein Tau as detected by chemical cross- linking and phosphopeptide mapping. J. Biol. Chem 1999, 274, 8029–8038. [Google Scholar]
- Sibille, N.; Sillen, A.; Leroy, A.; Wieruszeski, J.M.; Mulloy, B.; Landrieu, I.; Lippens, G. Structural impact of heparin binding to full- length Tau as studied by NMR spectroscopy. Biochemistry 2006, 45, 12560–12572. [Google Scholar]
- Ramachandran, G.; Udgaonkar, J.B. Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by Tau protein. J. Biol. Chem 2011, 286, 38948–38959. [Google Scholar]
- Ramachandran, G.; Udgaonkar, J.B. Evidence for the existence of a secondary pathway for fibril growth during the aggregation of Tau. J. Mol. Biol 2012, 421, 296–314. [Google Scholar]
- Elbaum-Garfinkle, S.; Rhoades, E. Identification of an aggregation-prone structure of Tau. J. Am. Chem. Soc 2012, 134, 16607–16613. [Google Scholar]
- Bocharova, O.V.; Breydo, L.; Parfenov, A.S.; Salnikov, V.V.; Baskakov, I.V. In vitro conversion of full- length mammalian prion protein produces amyloid form with physical properties of PrP Sc. J. Mol. Biol 2005, 346, 645–659. [Google Scholar]
- Makarava, N.; Lee, C.I.; Ostapchenko, V.G.; Baskakov, I.V. Highly promiscuous nature of prion polymerization. J. Biol. Chem 2007, 282, 36704–36713. [Google Scholar]
- Zhou, B.R.; Zhou, Z.; Hu, Q.L.; Chen, J.; Liang, Y. Mixed macromolecular crowding inhibits amyloid formation of hen egg white lysozyme. Biochim. Biophys. Acta 2008, 1784, 472–480. [Google Scholar]
- Krebs, M.R.; Wilkins, D.K.; Chung, E.W.; Pitkeathly, M.C.; Chamberlain, A.K.; Zurdo, J.; Robinson, C.V.; Dobson, C.M. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain. J. Mol. Biol 2000, 300, 541–549. [Google Scholar]
- Jiao, M.; Li, H.T.; Chen, J.; Minton, A.P.; Liang, Y. Attractive protein-polymer interactions markedly alter the effect of macromolecular crowding on protein association equilibria. Biophys. J 2010, 99, 914–923. [Google Scholar]
- Minton, A.P. Quantitative assessment of the relative contributions of steric repulsion and chemical interactions to macromolecular crowding. Biopolymers 2013, 99, 239–244. [Google Scholar]
- Vagenende, V.; Han, A.X.; Pek, H.B.; Loo, B.L. Quantifying the molecular origins of opposite solvent effects on protein-protein interactions. PloS Comput. Biol 2013, 9, e1003072. [Google Scholar]
- Rosen, J.; Kim, Y.C.; Mittal, J. Modest protein-crowder attractive interactions can counteract enhancement of protein association by intermolecular excluded volume interactions. J. Phys. Chem. B 2011, 115, 2683–2689. [Google Scholar]
- Zhang, D.L.; Wu, L.J.; Chen, J.; Liang, Y. Effects of macromolecular crowding on the structural stability of human α- lactalbumin. Acta Biochim. Biophys. Sin 2012, 44, 703–711. [Google Scholar]
- Seeliger, J.; Werkmüller, A.; Winter, R. Macromolecular crowding as a suppressor of human IAPP fibril formation and cytotoxicity. PloS One 2013, 8, e69652. [Google Scholar]
- Finn, T.E.; Nunez, A.C.; Sunde, M.; Easterbrook-Smith, S.B. Serum albumin prevents protein aggregation and amyloid formation and retains chaperone- like activity in the presence of physiological ligands. J. Biol. Chem 2012, 287, 21530–21540. [Google Scholar]
- Zhou, B.R.; Liang, Y.; Du, F.; Zhou, Z.; Chen, J. Mixed macromolecular crowding accelerates the oxidative refolding of reduced, denatured lysozyme: Implications for protein folding in intracellular environments. J. Biol. Chem 2004, 279, 55109–55116. [Google Scholar]
- Minton, A.P. The effective hard particle model provides a simple, robust, and broadly applicable description of nonideal behavior in concentrated solutions of bovine serum albumin and other nonassociating proteins. J. Pharm. Sci 2007, 96, 3466–3469. [Google Scholar]
- Wang, Y.; Sarkar, M.; Smith, A.E.; Krois, A.S.; Pielak, G.J. Macromolecular crowding and protein stability. J. Am. Chem. Soc 2012, 134, 16614–16618. [Google Scholar]
- Feig, M.; Sugita, Y. Variable interactions between protein crowders and biomolecular solutes are important in understanding cellular crowding. J. Phys. Chem. B 2012, 116, 599–605. [Google Scholar]
- Endo, T.; Groth, D.; Prusiner, S.B.; Kobata, A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 1989, 28, 8380–8388. [Google Scholar]
- Rudd, P.M.; Wormald, M.R.; Wing, D.R.; Prusiner, S.B.; Dwek, R.A. Prion glycoprotein: Structure, dynamics, and roles for the sugars. Biochemistry 2001, 40, 3759–3766. [Google Scholar]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar]
- Huang, L.; Jin, R.; Li, J.; Luo, K.; Huang, T.; Wu, D.; Wang, W.; Chen, R.; Xiao, G.F. Macromolecular crowding converts the human recombinant PrPC to the soluble neurotoxic β-oligomers. FASEB J 2010, 24, 3536–3543. [Google Scholar]
- Bergasa-Caceres, F.; Rabitz, H.A. A simple quantitative model of macromolecular crowding effects on protein folding: Application to the murine prion protein (121–231). Chem. Phys. Lett 2013, 574, 112–115. [Google Scholar]
- Vorberg, I.; Groschup, M.H.; Pfaff, E.; Priola, S.A. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J. Virol 2003, 77, 2003–2009. [Google Scholar]
- Prusiner, S.B. Unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar]
- Soto, C. Transmissible proteins: Expanding the prion heresy. Cell 2012, 149, 968–977. [Google Scholar]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell. Biol 2009, 11, 909–913. [Google Scholar]
- Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Accelerated fibrillation of α-synuclein induced by the combined action of macromolecular crowding and factors inducing partial folding. Curr. Alzheimer Res 2009, 6, 252–260. [Google Scholar]
- Stathopulos, P.B.; Rumfeldt, J.A.; Scholz, G.A.; Irani, R.A.; Frey, H.E.; Hallewell, R.A.; Lepock, J.R.; Meiering, E.M. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 7021–7026. [Google Scholar]
- Li, C.; Xu, W.C.; Xie, Z.S.; Pan, K.; Hu, J.; Chen, J.; Pang, D.W.; Yang, F.Q.; Liang, Y. Cupric ions induce the oxidation and trigger the aggregation of human superoxide dismutase 1. PloS One 2013, 8, e65287. [Google Scholar]
- Vassall, K.A.; Stubbs, H.R.; Primmer, H.A.; Tong, M.S.; Sullivan, S.M.; Sobering, R.; Srinivasan, S.; Briere, L.-A.K.; Dunn, S.D.; Colón, W.; et al. Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc. Natl. Acad. Sci. USA 2011, 108, 2210–2215. [Google Scholar]
- Yeung, P.S.; Axelsen, P.H. The crowded environment of a reverse micelle induces the formation of β-strand seed structures for nucleating amyloid fibril formation. J. Am. Chem. Soc 2012, 134, 6061–6063. [Google Scholar]
- Bokvist, M.; Grobner, G. Misfolding of amyloidogenic proteins at membrane surfaces: The impact of macromolecular crowding. J. Am. Chem. Soc 2007, 129, 14848–14849. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar]
- Ellis, R.J.; Minton, A.P. Cell biology: Join the crowd. Nature 2003, 425, 27–28. [Google Scholar]
- Engel, R.; Westphal, A.H.; Huberts, D.H.; Nabuurs, S.M.; Lindhoud, S.; Visser, A.J.; van Mierlo, C.P. Macromolecular crowding compacts unfolded apoflavodoxin and causes severe aggregation of the off-pathway intermediate during apoflavodoxin folding. J. Biol. Chem 2008, 283, 27383–27394. [Google Scholar]
- Du, F.; Zhou, Z.; Mo, Z.Y.; Shi, J.Z.; Chen, J.; Liang, Y. Mixed macromolecular crowding accelerates the refolding of rabbit muscle creatine kinase: Implications for protein folding in physiological environments. J. Mol. Biol 2006, 364, 469–482. [Google Scholar]
- Hatters, D.M.; Minton, A.P.; Howlett, G.J. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J. Biol. Chem 2002, 277, 7824–7830. [Google Scholar]
- Coquel, A.S.; Jacob, J.P.; Primet, M.; Demarez, A.; Dimiccoli, M.; Julou, T.; Moisan, L.; Lindner, A.B.; Berry, H. Localization of protein aggregation in Escherichia coli is governed by diffusion and nucleoid macromolecular crowding effect. PloS Comput. Biol 2013, 9, e1003038. [Google Scholar]
- Milles, S.; Huy Bui, K.; Koehler, C.; Eltsov, M.; Beck, M.; Lemke, E.A. Facilitated aggregation of FG nucleoporins under molecular crowding conditions. EMBO Rep 2013, 14, 178–183. [Google Scholar]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci 2003, 4, 49–60. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, Q.; Hu, J.-Y.; Chen, J.; Liang, Y. The Role of Crowded Physiological Environments in Prion and Prion-like Protein Aggregation. Int. J. Mol. Sci. 2013, 14, 21339-21352. https://doi.org/10.3390/ijms141121339
Ma Q, Hu J-Y, Chen J, Liang Y. The Role of Crowded Physiological Environments in Prion and Prion-like Protein Aggregation. International Journal of Molecular Sciences. 2013; 14(11):21339-21352. https://doi.org/10.3390/ijms141121339
Chicago/Turabian StyleMa, Qian, Ji-Ying Hu, Jie Chen, and Yi Liang. 2013. "The Role of Crowded Physiological Environments in Prion and Prion-like Protein Aggregation" International Journal of Molecular Sciences 14, no. 11: 21339-21352. https://doi.org/10.3390/ijms141121339
APA StyleMa, Q., Hu, J. -Y., Chen, J., & Liang, Y. (2013). The Role of Crowded Physiological Environments in Prion and Prion-like Protein Aggregation. International Journal of Molecular Sciences, 14(11), 21339-21352. https://doi.org/10.3390/ijms141121339