Opportunities for Live Cell FT-Infrared Imaging: Macromolecule Identification with 2D and 3D Localization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. IR Absorption Spectroscopy
1.2. IR Spectrometers, Sources, and Detectors
1.3. Synchrotron Radiation FTIR (SR-FTIR) Raster Scanning and Widefield Spectromicroscopy
2. Experimental and Analytical Methods
2.1. 2D Projection Raster Scanning and Widefield Spectromicroscopy
2.1.1. Transmission
2.1.2. Transflection
2.2. 3D IR Spectral Microtomography
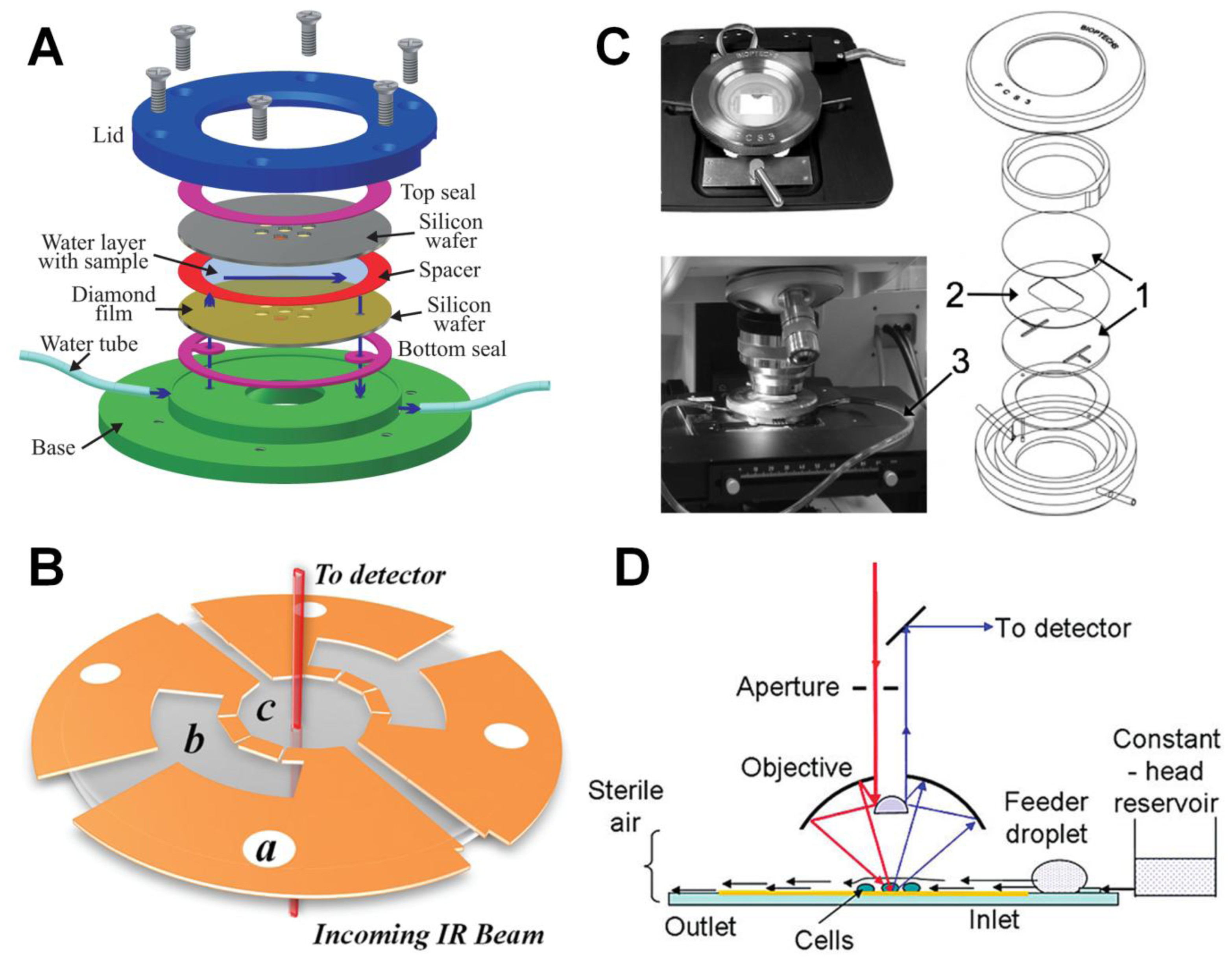
2.3. Microfluidics
2.4. Scattering Effects
2.5. Large Data Sets
2.6. Diffraction-Limited Spatial Resolution
3. Examples
3.1. 2D Projection RS and WF FTIR Spectromicroscopy
3.1.1. Transmission
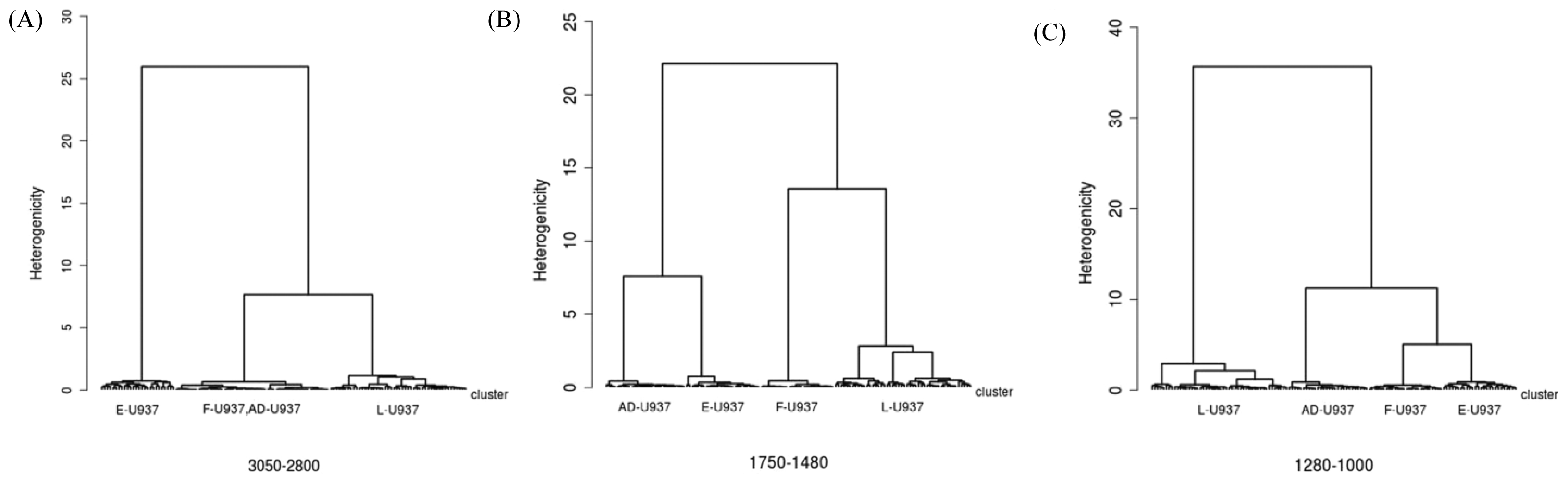
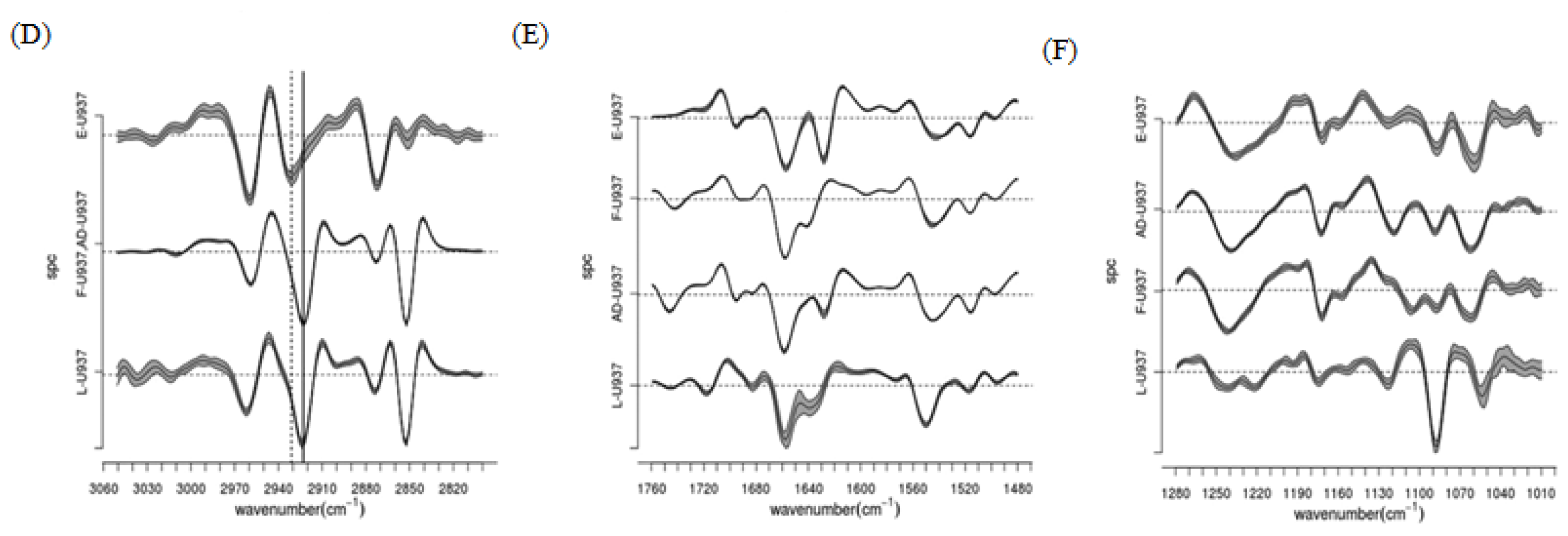
3.1.1.1. Biochemical Alterations Induced by Different Fixation Protocols on U937 Leukemic Monocytes (RS Spectromicroscopy)
3.1.1.2. Arsenic-Induced Changes to Intracellular Biomolecules in Live Leukemia Cells (RS Spectromicroscopy)
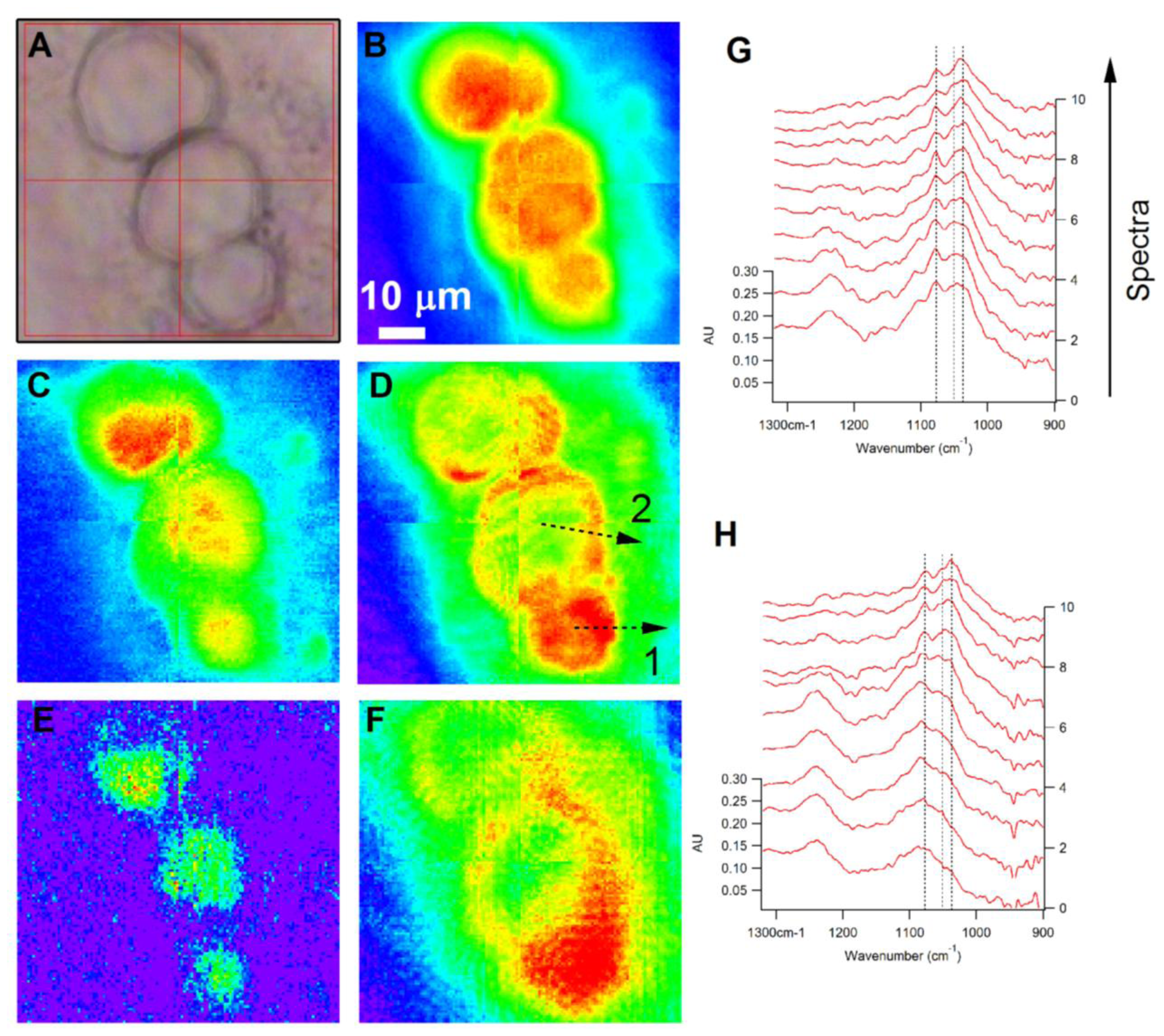
3.1.1.3. Subcellular Imaging of Chemical Moieties in Sensory Neurons (WF FTIR Spectromicroscopy)
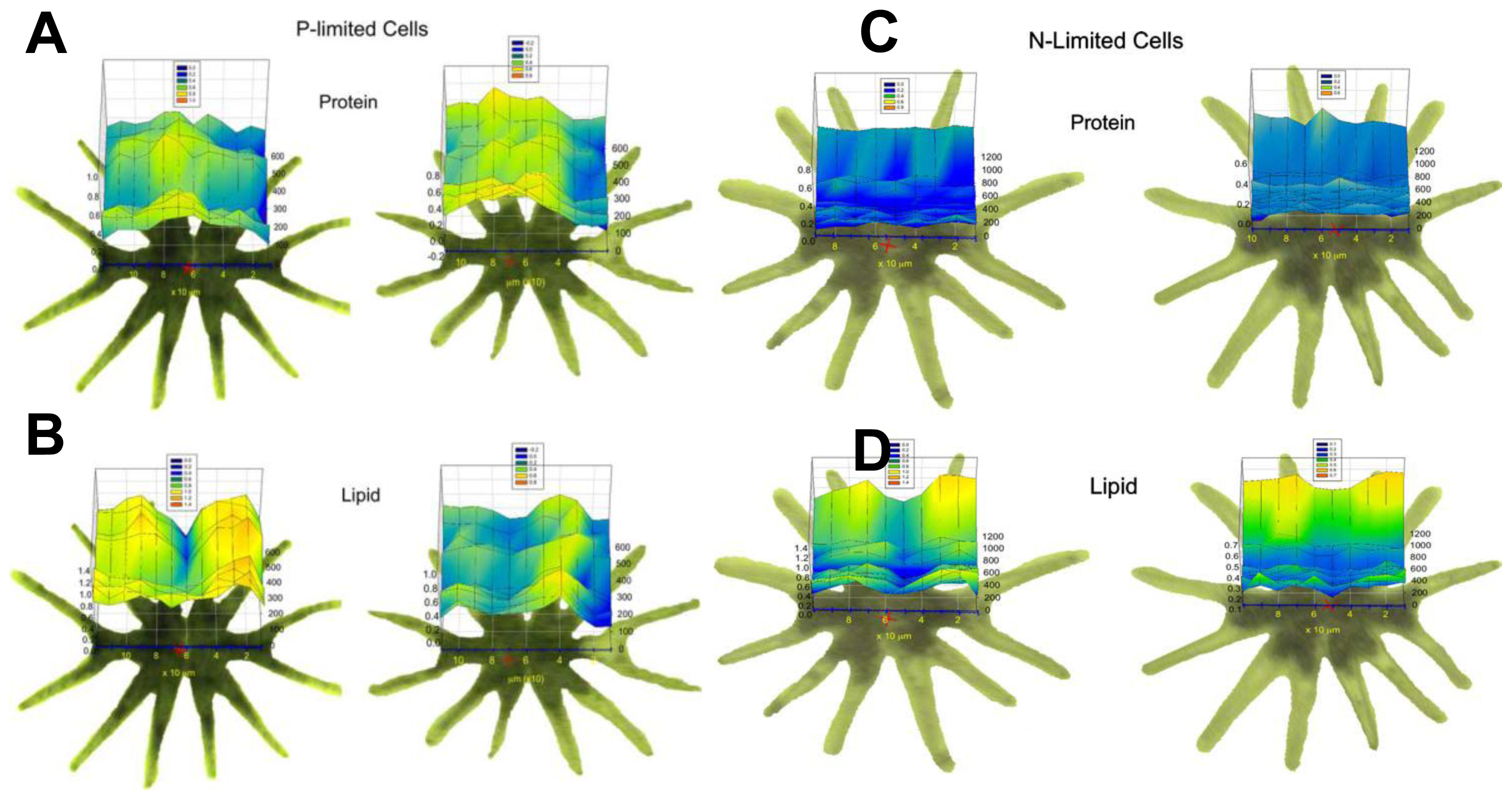
3.1.1.4. Time Dependent Macromolecule Changes in Micrasterias Due to Nitrogen Source (RS FTIR Spectromicroscopy)
3.1.1.5. Time Dependent Macromolecule Changes in Thallasosira Weissflogii Due to Elevated Carbon Dixide Exposure (WF FTIR Spectromicroscopy)
3.1.2. Transflection
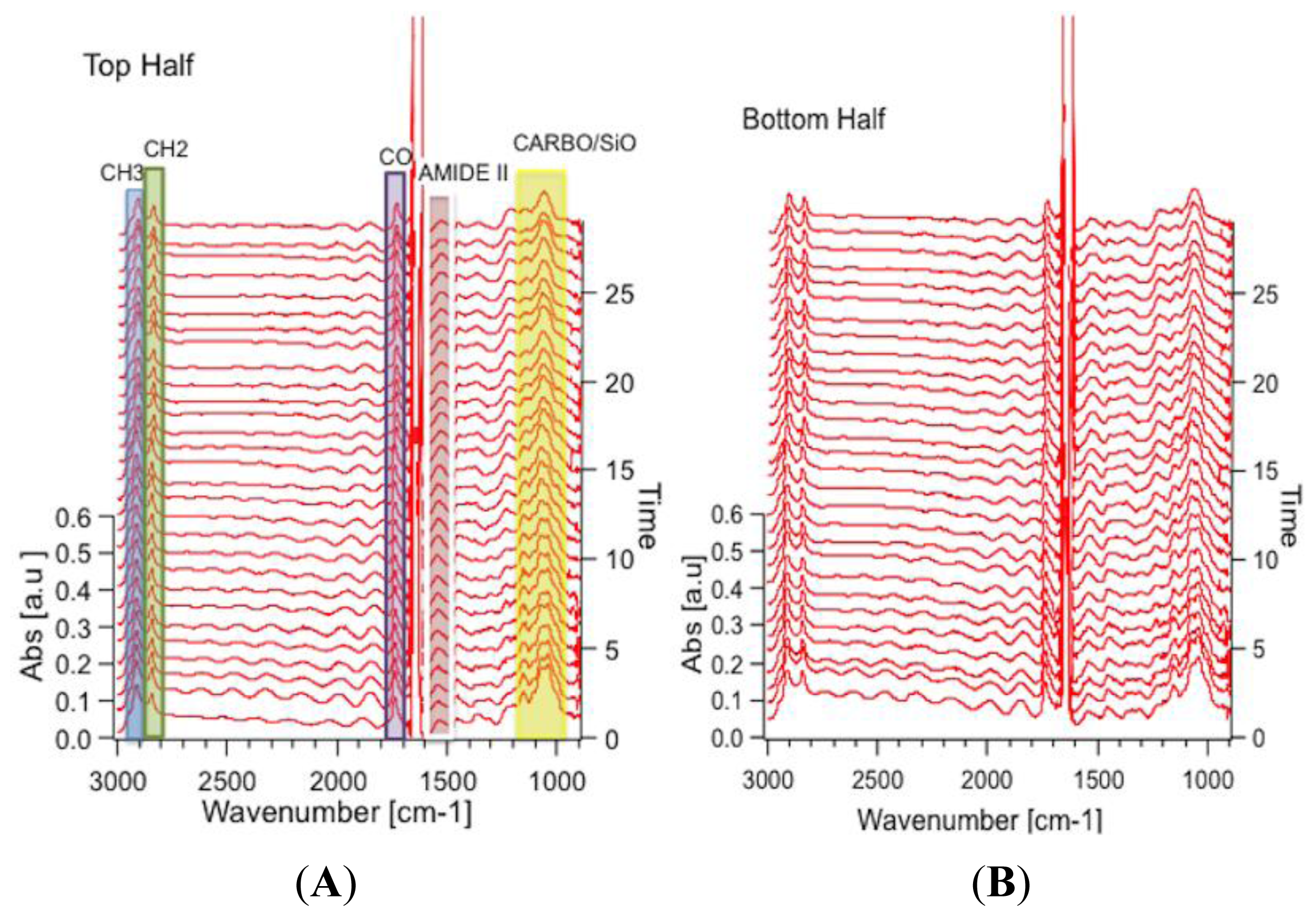
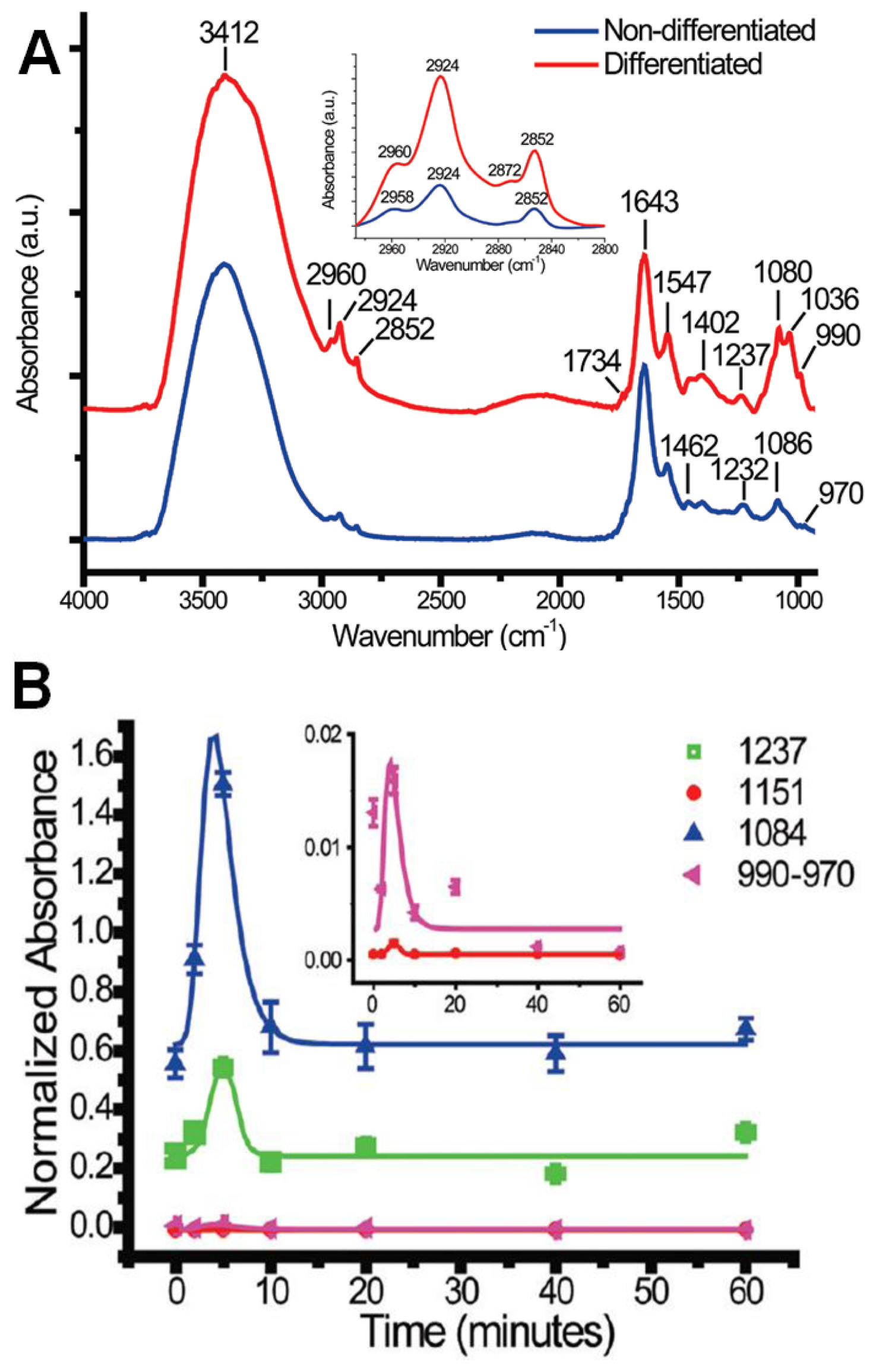
3.1.2.1. Protein Phosphorylation in Single PC12 Cells during Neuronal Differentiation (RS FTIR Spectromicroscopy)
3.2. 3D Spectral Microtomography
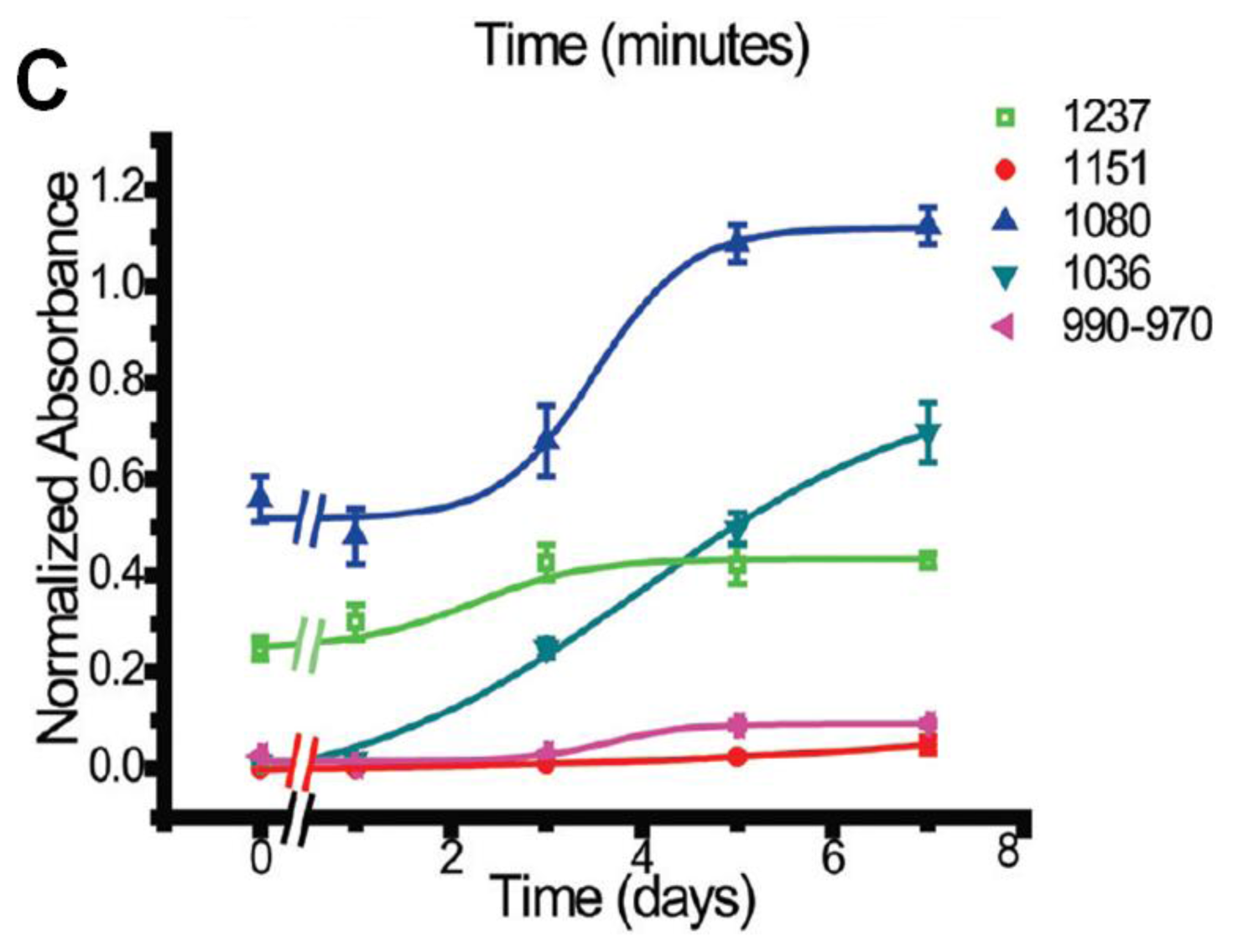
3.2.1. Macromolecular Architecture of a Colony of Stem Cells
4. Conclusions











Conflicts of Interest
References
- Martin, M.C.; Schade, U.; Lerch, P.; Dumas, P. Recent applications and current trends in analytical chemistry using synchrotron-based Fourier-transform infrared microspectroscopy. Trends Anal. Chem 2010, 29, 453–463. [Google Scholar]
- Hirschmugl, C.J.; Gough, K.M. Fourier transform infrared spectrochemical imaging: Review of design and applications with a focal plane array and multiple beam synchrotron radiation source. Appl. Spectrosc 2012, 66, 475–491. [Google Scholar]
- Nasse, M.J.; Ratti, S.; Giordano, M.; Hirschmugl, C.J. Demountable liquid/flow cell for in vivo infrared microspectroscopy of biological specimens. Appl. Spectrosc 2009, 63, 1181–1186. [Google Scholar]
- Tobin, M.J.; Puskar, L.; Hasan, J.; Webb, H.K.; Hirschmugl, C.J.; Nasse, M.J.; Gervinskas, G.; Juodkazis, S.; Watson, G.S.; Watson, J.A.; et al. High-spatial-resolution mapping of superhydro-phobic cicada wing surface chemistry using infrared microspectroscopy and infrared imaging at two synchrotron beamlines. J. Synchrot. Radiat 2013, 20, 482–489. [Google Scholar]
- Holman, H.Y.N.; Miles, R.; Hao, Z.; Wozei, E.; Anderson, L.M.; Yang, H. Real-time chemical imaging of bacterial activity in biofilms using open-channel microfluidics and synchrotron FTIR spectromicroscopy. Anal. Chem 2009, 81, 8564–8570. [Google Scholar]
- Vaccari, L.; Birarda, G.; Businaro, L.; Pacor, S.; Grenci, G. Infrared microspectroscopy of live cells in microfluidic devices (MD-IRMS): Toward a powerful label-free cell-based assay. Anal. Chem 2012, 84, 4768–4775. [Google Scholar]
- Martin, M.C.; Dabat-Blondeau, C.; Unger, M.; Sedlmair, J.; Parkinson, D.Y.; Bechtel, H.A.; Illman, B.; Castro, J.M.; Keiluweit, M.; Buschke, D.; et al. 3D spectral imaging with synchrotron Fourier transform infrared spectro-microtomography. Nat. Methods 2013, 10, 861–864. [Google Scholar]
- Whelan, D.R.; Bambery, K.R.; Puskar, L.; McNaughton, D.; Wood, B.R. Synchrotron Fourier transform infrared (FTIR) analysis of single living cells progressing through the cell cycle. Analyst 2013, 138, 3891–3899. [Google Scholar]
- Heraud, P.; Wood, B.R.; Tobin, M.J.; Beardall, J.; McNaughton, D. Mapping of nutrient-induced biochemical changes in living algal cells using synchrotron infrared microspectroscopy. FEMS Microbiol. Lett 2005, 249, 219–225. [Google Scholar]
- Quaroni, L.; Zlateva, T. Infrared spectromicroscopy of biochemistry in functional single cells. Analyst 2011, 136, 3219–3232. [Google Scholar]
- Holman, H.Y.N.; Bechtel, H.A.; Hao, Z.; Martin, M.C. Synchrotron IR spectromicroscopy: Chemistry of living cells. Anal. Chem 2010, 82, 8757–8765. [Google Scholar]
- Meltzer, M. Albert Einstein: A Biography; Holiday House: New York, NY, USA, 2008. [Google Scholar]
- Griffiths, P.; de Haseth, J.A. Fourier Transform Infrared Spectrometry; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Tobin, M.J.; Puskar, L.; Barber, R.L.; Harvey, E.C.; Heraud, P.; Wood, B.R.; Bambery, K.R.; Dillon, C.T.; Munro, K.L. FTIR spectroscopy of single live cells in aqueous media by synchrotron IR microscopy using microfabricated sample holders. Vib. Spectrosc 2010, 53, 34–38. [Google Scholar]
- Siesler, H.W.; Salzer, R. Infrared and Raman Spectroscopic Imaging; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009. [Google Scholar]
- Pereira, M.F.; Shulika, O. Terahertz and Mid Infrared Radiation: Generation, Detection, and Applications, 1st ed; Springer: AA Dordrecht, The Netherlands, 2011. [Google Scholar]
- Faist, J.; Capasso, F.; Sivco, D.L.; Sirtori, C.; Hutchinson, A.L.; Cho, A.Y. Quantum cascade laser. Science 1994, 264, 553–556. [Google Scholar]
- Levin, I.W.; Bhargava, R. Fourier Transform Infrared Vibrational Spectroscopic Imaging: Integrating Microscopy and Molecular Recognition. In Annual Review of Physical Chemistry; Annual Reviews: Palo Alto, CA, USA, 2005; Volume 56, pp. 429–474. [Google Scholar]
- Reffner, J.A.; Martoglio, P.A.; Williams, G.P. Fourier-transform infrared microscopic analysis with synchrotron-radiation—The microscope optics and system performance. Rev. Sci. Instrum 1995, 66, 1298–1302. [Google Scholar]
- Carter, M.R.; Bennett, C.L.; Fields, D.J.; Lee, F.D. Livermore Imaging Fourier Transform Infrared Spectrometer (LIFTIRS), Proc. SPIE 2480, Orlando, FL, USA; 1995; pp. 380–386.
- Moss, D.; Gasharova, B.; Mathis, Y.L. Practical tests of a focal plane array detector microscope at the ANKA-IR beamline. Infrared Phys. Technol 2006, 49, 53–56. [Google Scholar]
- Carr, G.L.; Chubar, O.; Dumas, P. Multichannel Detection with a Synchrotron Light Source: Design and Potential. In Spectrochemical Analysis Using Infrared Multichannel Detectors; Blackwell Publishing Ltd: Urbana, IL, USA, 2007; pp. 56–84. [Google Scholar]
- Miller, L.M.; Dumas, P. Chemical imaging of biological tissue with synchrotron infrared light. Biochim. Biophys. Acta Biomembr 2006, 1758, 846–857. [Google Scholar]
- Nasse, M.J.; Walsh, M.J.; Mattson, E.C.; Reininger, R.; Kajdacsy-Balla, A.; Macias, V.; Bhargava, R.; Hirschmugl, C.J. High-resolution Fourier-transform infrared chemical imaging with multiple synchrotron beams. Nat. Methods 2011, 8, 413–416. [Google Scholar]
- Mass, J.; Sedlmair, J.; Patterson, C.S.; Carson, D.; Buckley, B.; Hirschmugl, C. SR-FTIR imaging of the altered cadmium sulfide yellow paints in Henri Matisse’s Le Bonheur de vivre 1905–6—Examination of visually distinct degradation regions. Analyst 2013, 138, 6032–6043. [Google Scholar]
- Clede, S.; Lambert, F.; Sandt, C.; Kascakova, S.; Unger, M.; Harte, E.; Plamont, M.-A.; Saint-Fort, R.; Deniset-Besseau, A.; Gueroui, Z.; et al. Detection of an estrogen derivative in two breast cancer cell lines using a single core multimodal probe for imaging (SCoMPI) imaged by a panel of luminescent and vibrational techniques. Analyst 2013, 138, 5627–5638. [Google Scholar]
- Clemons, C.; Sedlmair, J.; Illman, B.; Ibach, R.; Hirschmugl, C. Chemically imaging the effects of the addition of nanofibrillated cellulose on the distribution of poly(acrylic acid) in poly(vinyl alcohol). Polymer 2013, 54, 2058–2061. [Google Scholar]
- Hackett, M.J.; Borondics, F.; Brown, D.; Hirschmugl, C.; Smith, S.E.; Paterson, P.G.; Nichol, H.; Pickering, I.J.; George, G.N. Subcellular biochemical investigation of purkinje neurons using synchrotron radiation fourier transform infrared spectroscopic imaging with a focal plane array detector. ACS Chem. Neurosci 2013, 4, 1071–1080. [Google Scholar]
- Liao, C.R.; Rak, M.; Lund, J.; Unger, M.; Platt, E.; Albensi, B.C.; Hirschmugl, C.J.; Gough, K.M. Synchrotron FTIR reveals lipid around and within amyloid plaques in transgenic mice and Alzheimer’s disease brain. Analyst 2013, 138, 3991–3997. [Google Scholar]
- Mattson, E.C.; Unger, M.; Clede, S.; Lambert, F.; Policar, C.; Imtiaz, A.; D’Souza, R.; Hirschmugl, C.J. Toward optimal spatial and spectral quality in widefield infrared spectromicroscopy of IR labelled single cells. Analyst 2013, 138, 5610–5618. [Google Scholar]
- Mattson, E.C.; Pande, K.; Unger, M.; Cui, S.M.; Lu, G.H.; Gajdardziska-Josifovska, M.; Weinert, M.; Chen, J.H.; Hirschmugl, C.J. Exploring adsorption and reactivity of NH3 on reduced graphene oxide. J. Phys. Chem. C 2013, 117, 10698–10707. [Google Scholar]
- Fogarty, S.W.; Patel, I.I.; Trevisan, J.; Nakamura, T.; Hirschmugl, C.J.; Fullwood, N.J.; Martin, F.L. Sub-cellular spectrochemical imaging of isolated human corneal cells employing synchrotron radiation-based Fourier-transform infrared microspectroscopy. Analyst 2013, 138, 240–248. [Google Scholar]
- Unger, M.; Mattson, E.; Patterson, C.S.; Alavi, Z.; Carson, D.; Hirschmugl, C.J. Synchrotron-based multiple-beam FTIR chemical imaging of a multi-layered polymer in transmission and reflection: Towards cultural heritage applications. Appl. Phys. A Mater. Sci. Process 2013, 111, 135–145. [Google Scholar]
- Riding, M.J.; Trevisan, J.; Hirschmugl, C.J.; Jones, K.C.; Semple, K.T.; Martin, F.L. Mechanistic insights into nanotoxicity determined by synchrotron radiation-based Fouriertransform infrared imaging and multivariate analysis. Environ. Int 2012, 50, 56–65. [Google Scholar]
- Schmidt-Patterson, C.; Carson, D.; Phenix, A.; Khanjian, H.; Trentelman, K.; Mass, J.; Hirschmugl, C.J. Synchrotron-based imaging FTIR spectroscopy in the evaluation of painting cross-sections. E Preserv. Sci 2013, 10, 1–9. [Google Scholar]
- Mattson, E.C.; Nasse, M.J.; Rak, M.; Gough, K.M.; Hirschmugl, C.J. Restoration and spectral recovery of mid-infrared chemical images. Anal. Chem 2012, 84, 6173–6180. [Google Scholar]
- Kastyak-Ibrahim, M.Z.; Nasse, M.J.; Rak, M.; Hirschmugl, C.; del Bigio, M.R.; Albensi, B.C.; Gough, K.M. Biochemical label-free tissue imaging with subcellular-resolution synchrotron FTIR with focal plane array detector. Neuroimage 2012, 60, 376–383. [Google Scholar]
- Nasse, M.J.; Bellehumeur, B.; Ratti, S.; Olivieri, C.; Buschke, D.; Squirrell, J.; Eliceiri, K.; Ogle, B.; Patterson, C.S.; Giordano, M.; et al. Opportunities for multiple-beam synchrotron-based mid-infrared imaging at IRENI. Vib. Spectrosc 2012, 60, 10–15. [Google Scholar]
- Stuart, B.H. Infrared Spectroscopy: Fundamentals and Applications; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Heraud, P.; Stojkovic, S.; Beardall, J.; McNaughton, D.; Wood, B.R. Intercolonial variability in macromolecular composition in P-starved and P-replete scenedesmus populations revealed by infrared microspectroscopy. J. Phycol 2008, 44, 1335–1339. [Google Scholar]
- Birarda, G.; Grenci, G.; Businaro, L.; Marmiroli, B.; Pacor, S.; Piccirilli, F.; Vaccari, L. Infrared microspectroscopy of biochemical response of living cells in microfabricated devices. Vib. Spectrosc 2010, 53, 6–11. [Google Scholar]
- Birarda, G.; Grenci, G.; Businaro, L.; Marmiroli, B.; Pacor, S.; Vaccari, L. Fabrication of a microfluidic platform for investigating dynamic biochemical processes in living samples by FTIR microspectroscopy. Microelectron. Eng 2010, 87, 806–809. [Google Scholar]
- Filik, J.; Frogley, M.D.; Pijanka, J.K.; Wehbe, K.; Cinque, G. Electric field standing wave artefacts in FTIR micro-spectroscopy of biological materials. Analyst 2012, 137, 853–861. [Google Scholar]
- Bassan, P.; Lee, J.; Sachdeva, A.; Pissardini, J.; Dorling, K.M.; Fletcher, J.S.; Henderson, A.; Gardner, P. The inherent problem of transflection-mode infrared spectroscopic microscopy and the ramifications for biomedical single point and imaging applications. Analyst 2013, 138, 144–157. [Google Scholar]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol 2007, 8, 839–845. [Google Scholar]
- Herman, G.T. Fundamentals of Computerized Tomography: Image Reconstruction from Projection, 2nd ed; Springer: New York, NY, USA, 2010. [Google Scholar]
- Bassan, P.; Kohler, A.; Martens, H.; Lee, J.; Byrne, H.J.; Dumas, P.; Gazi, E.; Brown, M.; Clarke, N.; Gardner, P. Resonant Mie Scattering (RMieS) correction of infrared spectra from highly scattering biological samples. Analyst 2010, 135, 268–277. [Google Scholar]
- Bassan, P.; Byrne, H.J.; Lee, J.; Bonnier, F.; Clarke, C.; Dumas, P.; Gazi, E.; Brown, M.D.; Clarke, N.W.; Gardner, P. Reflection contributions to the dispersion artefact in FTIR spectra of single biological cells. Analyst 2009, 134, 1171–1175. [Google Scholar]
- Bassan, P.; Kohler, A.; Martens, H.; Lee, J.; Jackson, E.; Lockyer, N.; Dumas, P.; Brown, M.; Clarke, N.; Gardner, P. RMieS-EMSC correction for infrared spectra of biological cells: Extension using full Mie theory and GPU computing. J. Biophotonics 2010, 3, 609–620. [Google Scholar]
- Davis, B.J.; Carney, P.S.; Bhargava, R. Theory of mid-infrared absorption microspectroscopy. II: Heterogeneous samples. Anal. Chem 2010, 82, 3487–3499. [Google Scholar]
- Davis, B.J.; Carney, P.S.; Bhargava, R. Theory of midinfrared absorption microspectroscopy. I: Homogeneous samples. Anal. Chem 2010, 82, 3474–3486. [Google Scholar]
- Diem, M.; Papamarkakis, K.; Schubert, J.; Bird, B.; Romeo, M.J.; Miljkovic, M. The infrared spectral signatures of disease: Extracting the distinguishing spectral features between normal and diseased states. Appl. Spectrosc 2009, 63, 307A–318A. [Google Scholar]
- Bassan, P.; Sachdeva, A.; Kohler, A.; Hughes, C.; Henderson, A.; Boyle, J.; Shanks, J.H.; Brown, M.; Clarke, N.W.; Gardner, P. FTIR microscopy of biological cells and tissue: Data analysis using resonant Mie scattering (RMieS) EMSC algorithm. Analyst 2012, 137, 1370–1377. [Google Scholar]
- Bassan, P.; Byrne, H.J.; Bonnier, F.; Lee, J.; Dumas, P.; Gardner, P. Resonant Mie scattering in infrared spectroscopy of biological materials—Understanding the “dispersion artefact”. Analyst 2009, 134, 1586–1593. [Google Scholar]
- Lasch, P.; Diem, M.; Hansch, W.; Naumann, D. Artificial neural networks as supervised techniques for FT-IR microspectroscopic imaging. J. Chemometr 2006, 20, 209–220. [Google Scholar]
- Lasch, P.; Haensch, W.; Naumann, D.; Diem, M. Imaging of colorectal adenocarcinoma using FT-IR microspectroscopy and cluster analysis. Biochim. Biophys. Acta Mol. Basis Dis 2004, 1688, 176–186. [Google Scholar]
- Bhargava, R. Infrared spectroscopic imaging: The next generation. Appl. Spectrosc 2012, 66, 1091–1120. [Google Scholar]
- Prats-Montalban, J.M.; de Juan, A.; Ferrer, A. Multivariate image analysis: A review with applications. Chemometrics Intell. Lab. Syst 2011, 107, 1–23. [Google Scholar]
- Stelzer, E.H.K. Contrast, resolution, pixelation, dynamic range and signal-to-noise ratio: Fundamental limits to resolution in fluorescence light microscopy. J. Microsc. Oxf. 1998, 189, 15–24. [Google Scholar]
- Mattson, E.C.; Unger, M.; Manandhar, B.; Alavi, Z.; Hirschmugl, C.J. Multi-beam synchrotron FTIR chemical imaging: Impacts of schwarzschild objective and spatial oversampling on spatial resolution. J. Phys. Conf. Series 2013. [Google Scholar] [CrossRef]
- Carr, G.L. Resolution limits for infrared microspectroscopy explored with synchrotron radiation. Rev. Sci. Instrum 2001, 72, 1613–1619. [Google Scholar]
- Banyay, M.; Sandbrink, J.; Stromberg, R.; Graslund, A. Characterization of an RNA bulge structure by Fourier transform infrared spectroscopy. Biochem. Biophys. Res. Commun 2004, 324, 634–639. [Google Scholar]
- Banyay, M.; Sarkar, M.; Graslund, A. A library of IR bands of nucleic acids in solution. Biophys. Chem 2003, 104, 477–488. [Google Scholar]
- Bedolla, D.E.; Kenig, S.; Mitri, E.; Ferraris, P.; Marcello, A.; Grenci, G.; Vaccari, L. Determination of cell cycle phases in live B16 melanoma cells using IRMS. Analyst 2013, 138, 4015–4021. [Google Scholar]
- Lloyd, G.R.; Orr, L.E.; Christie-Brown, J.; McCarthy, K.; Rose, S.; Thomas, M.; Stone, N. Discrimination between benign, primary and secondary malignancies in lymph nodes from the head and neck utilising Raman spectroscopy and multivariate analysis. Analyst 2013, 138, 3900–3908. [Google Scholar]
- Patel, I.I.; Martin, F.L. Discrimination of zone-specific spectral signatures in normal human prostate using Raman spectroscopy. Analyst 2010, 135, 3060–3069. [Google Scholar]
- Munro, K.L.; Bambery, K.R.; Carter, E.A.; Puskar, L.; Tobin, M.J.; Wood, B.R.; Dillon, C.T. Synchrotron radiation infrared microspectroscopy of arsenic-induced changes to intracellular biomolecules in live leukemia cells. Vib. Spectrosc 2010, 53, 39–44. [Google Scholar]
- Munro, K.L.; Mariana, A.; Klavins, A.I.; Foster, A.J.; Lai, B.; Vogo, S.; Cai, Z.; Harris, H.H.; Dillon, C.T. Microprobe XRF mapping and XAS investigations of the intracellular metabolism of arsenic for understanding arsenic-induced toxicity. Chem. Res. Toxicol 2008, 21, 1760–1769. [Google Scholar]
- Johannes, C.B.; Le, T.K.; Zhou, X.L.; Johnston, J.A.; Dworkin, R.H. The prevalence of chronic pain in United States adults results of an internet-based survey. J. Pain 2010, 11, 1230–1239. [Google Scholar]
- Coderre, T.J.; Katz, J. Peripheral and central hyperexcitability: Differential signs and symptoms in persistent pain. Behav. Brain Sci 1997, 20, 404–419. [Google Scholar]
- Gracely, R.H.; Lynch, S.A.; Bennett, G.J. Painful neuropathy—Altered central processing maintained dynamically by peripheral input. Pain 1992, 51, 175–194. [Google Scholar]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar]
- Kuner, R. Central mechanisms of pathological pain. Nat. Med 2010, 16, 1258–1266. [Google Scholar]
- Dirajlal, S.; Pauers, L.E.; Stucky, C.L. Differential response properties of IB4-positive and -negative unmyelinated sensory neurons to protons and capsaicin. J. Neurophysiol 2003, 89, 513–524. [Google Scholar]
- Fullmer, J.M.; Riedl, M.S.; Higgins, L.A.; Elde, R. Identification of some lectin IB4 binding proteins in rat dorsal root ganglia. Neuroreport 2004, 15, 1705–1709. [Google Scholar]
- Gerke, M.B.; Plenderleith, M.B. Binding sites for the plant lectin Bandeiraea simplicifolia I-isolectin B4 are expressed by nociceptive primary sensory neurones. Brain Res 2001, 911, 101–104. [Google Scholar]
- Shelly, K.; Heraud, P.; Beardall, J. Nitrogen limitation in Dunaliella tertiolecta (Chlorophyceae) leads to increased susceptibility to damage by ultraviolet-B radiation but also increased repair capacity. J. Phycol 2002, 38, 713–720. [Google Scholar]
- Chen, L.; Holman, H.Y.N.; Hao, Z.; Bechtel, H.A.; Martin, M.C.; Wu, C.B.; Chu, S. Synchrotron infrared measurements of protein phosphorylation in living single PC12 cells during neuronal differentiation. Anal. Chem 2012, 84, 4118–4125. [Google Scholar]
- Fujita, K.; Lazarovici, P.; Guroff, G. Regulation of the differentiation of pc12 pheochromocytoma cells. Environ. Health Perspect 1989, 80, 127–142. [Google Scholar]
- Gomez, N.; Cohen, P. Dissection of the protein-kinase cascade by which nerve growth-factor activates map kinases. Nature 1991, 353, 170–173. [Google Scholar]
- Chang, J.H.; Mellon, E.; Schanen, N.C.; Twiss, J.L. Persistent TrkA activity is necessary to maintain transcription in neuronally differentiated PC12 cells. J. Biol. Chem 2003, 278, 42877–42885. [Google Scholar]
- Buschke, D.G.; Hei, D.J.; Eliceiri, K.W.; Ogle, B.M. Chapter 3 Screening Approaches for Stem Cells. In Stem Cell-Based Tissue Repair; The Royal Society of Chemistry: Cambridge, UK, 2011; pp. 45–80. [Google Scholar]
- Pijanka, J.K.; Kumar, D.; Dale, T.; Yousef, I.; Parkes, G.; Untereiner, V.; Yang, Y.; Dumas, P.; Collins, D.; Manfait, M.; et al. Vibrational spectroscopy differentiates between multipotent and pluripotent stem cells. Analyst 2010, 135, 3126–3132. [Google Scholar]
- Cao, J.; Ng, E.S.; McNaughton, D.; Stanley, E.G.; Elefanty, A.G.; Tobin, M.J.; Heraud, P. Fourier transform infrared microspectroscopy reveals unique phenotypes for human embryonic and induced pluripotent stem cell lines and their progeny. J Biophotonics 2013. [Google Scholar] [CrossRef]
- Ye, D.N.; Tanthanuch, W.; Thumanu, K.; Sangmalee, A.; Parnpai, R.; Heraud, P. Discrimination of functional hepatocytes derived from mesenchymal stem cells using FTIR microspectroscopy. Analyst 2012, 137, 4774–4784. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mattson, E.C.; Aboualizadeh, E.; Barabas, M.E.; Stucky, C.L.; Hirschmugl, C.J. Opportunities for Live Cell FT-Infrared Imaging: Macromolecule Identification with 2D and 3D Localization. Int. J. Mol. Sci. 2013, 14, 22753-22781. https://doi.org/10.3390/ijms141122753
Mattson EC, Aboualizadeh E, Barabas ME, Stucky CL, Hirschmugl CJ. Opportunities for Live Cell FT-Infrared Imaging: Macromolecule Identification with 2D and 3D Localization. International Journal of Molecular Sciences. 2013; 14(11):22753-22781. https://doi.org/10.3390/ijms141122753
Chicago/Turabian StyleMattson, Eric C., Ebrahim Aboualizadeh, Marie E. Barabas, Cheryl L. Stucky, and Carol J. Hirschmugl. 2013. "Opportunities for Live Cell FT-Infrared Imaging: Macromolecule Identification with 2D and 3D Localization" International Journal of Molecular Sciences 14, no. 11: 22753-22781. https://doi.org/10.3390/ijms141122753
APA StyleMattson, E. C., Aboualizadeh, E., Barabas, M. E., Stucky, C. L., & Hirschmugl, C. J. (2013). Opportunities for Live Cell FT-Infrared Imaging: Macromolecule Identification with 2D and 3D Localization. International Journal of Molecular Sciences, 14(11), 22753-22781. https://doi.org/10.3390/ijms141122753