Bone Morphogenetic Protein-7 Ameliorates Cerebral Ischemia and Reperfusion Injury via Inhibiting Oxidative Stress and Neuronal Apoptosis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
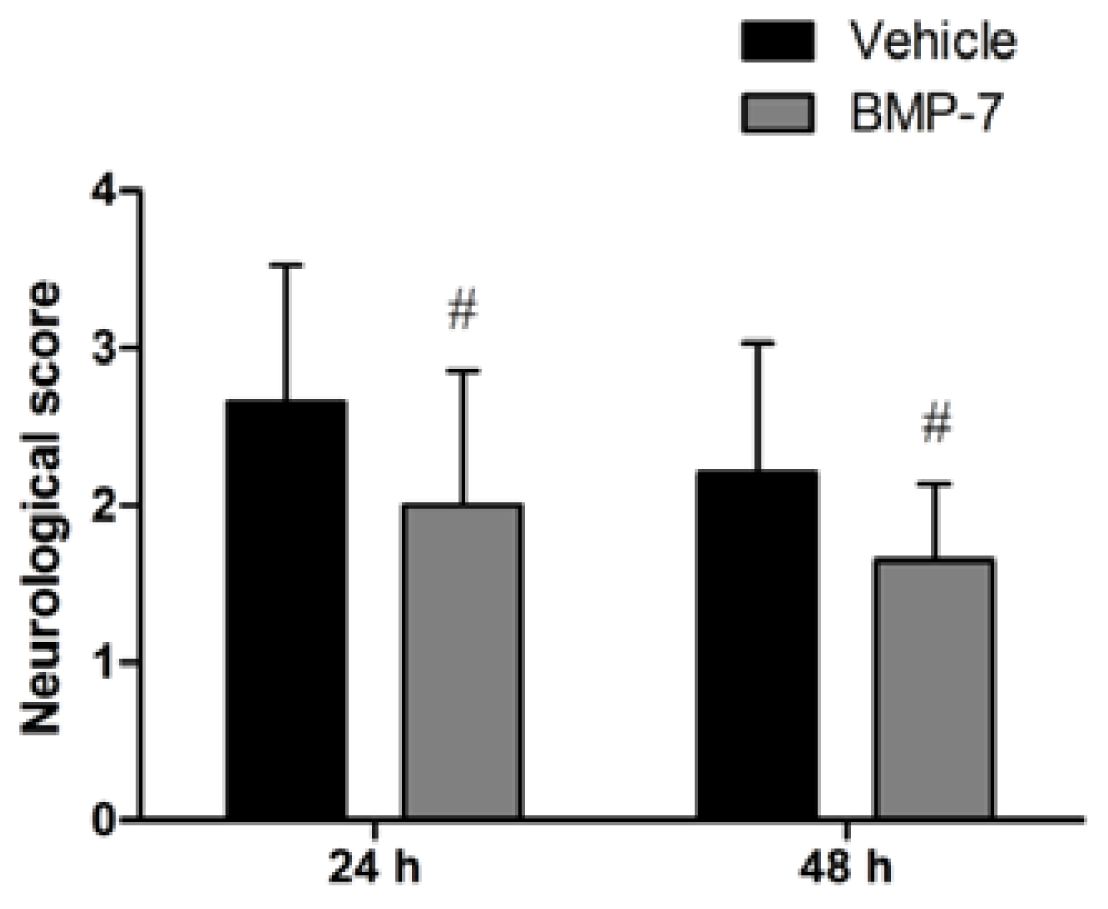
2.1. Neurological Deficits
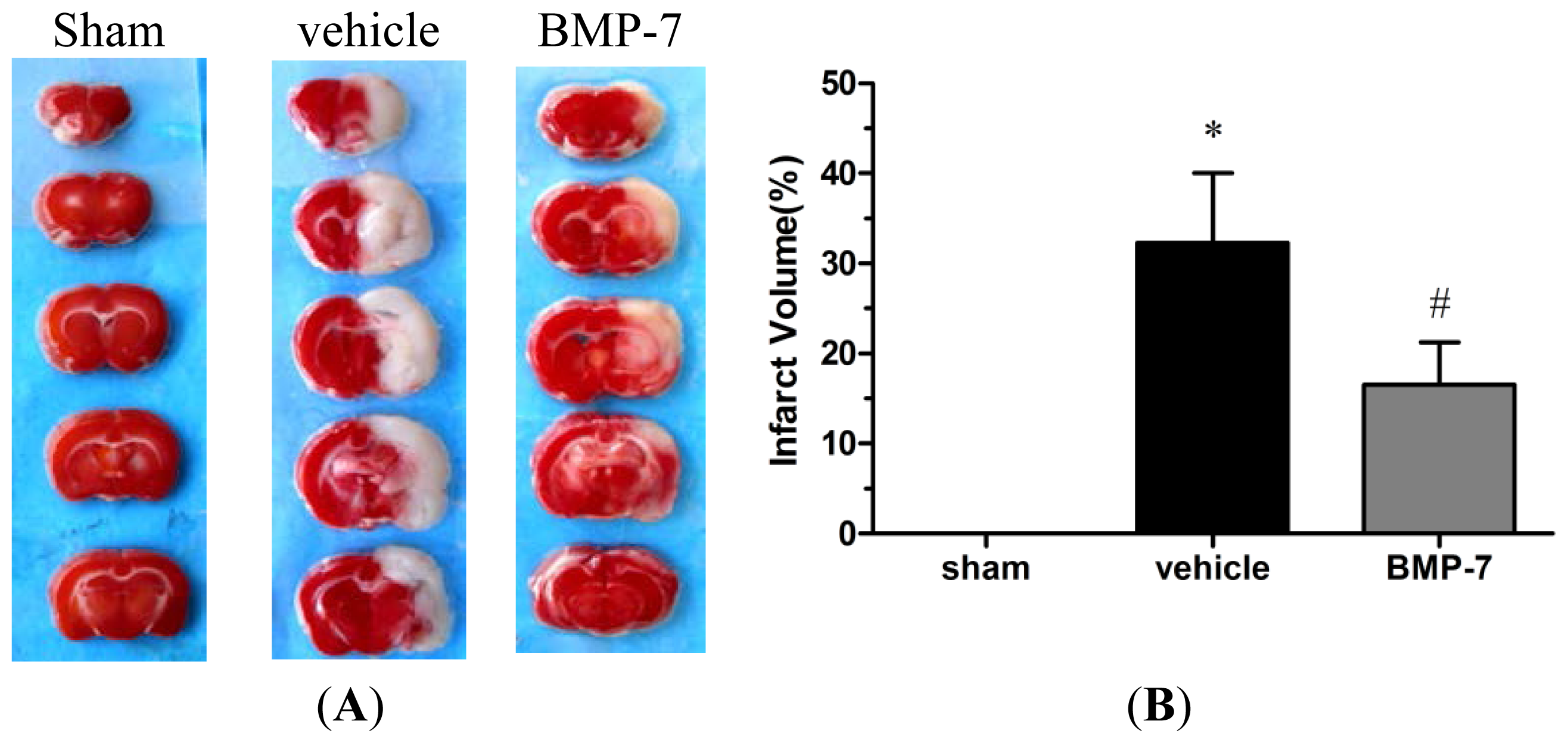
2.2. Effects of BMP-7 on Infarct Volume
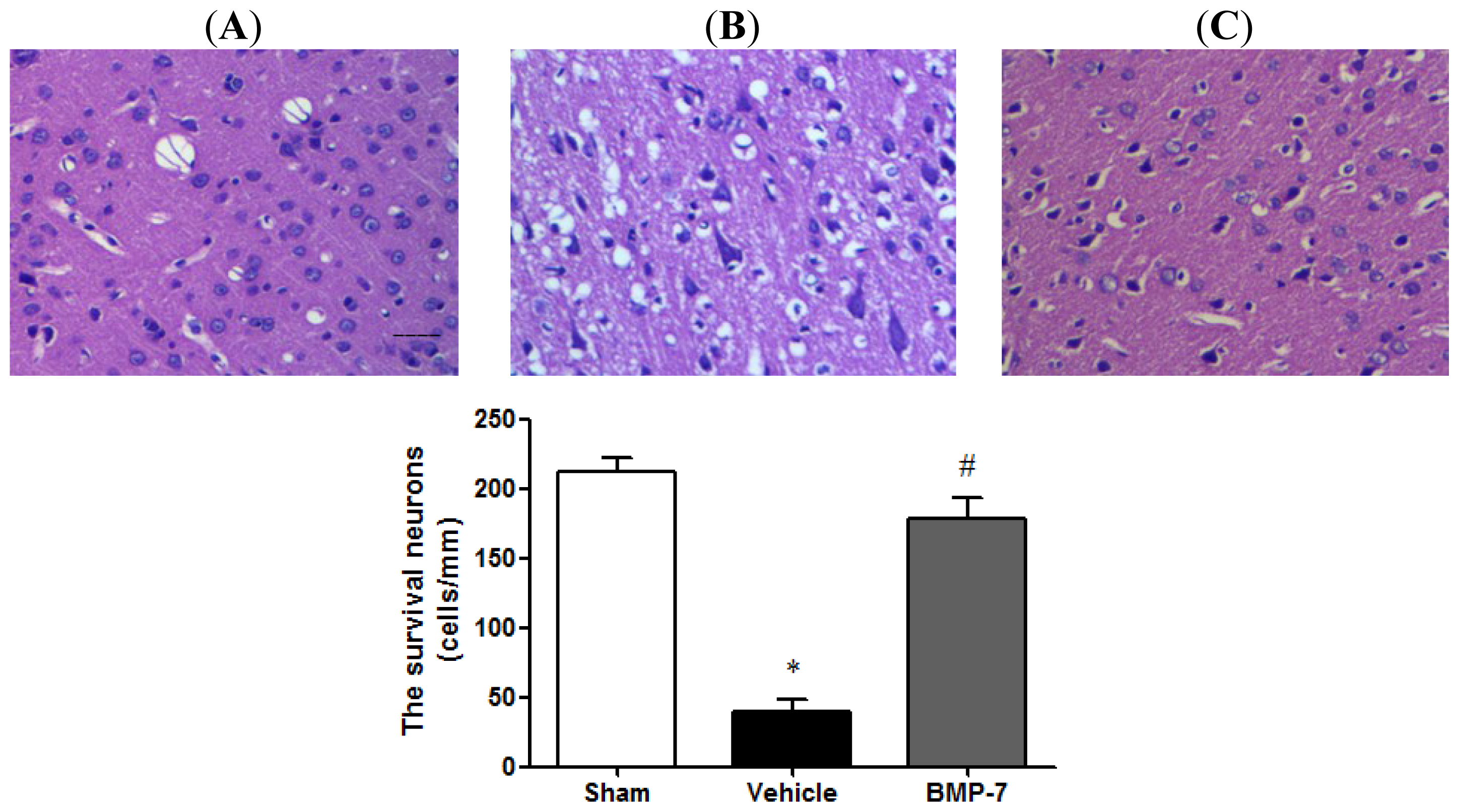
2.3. Effects of BMP-7 on Histopathology
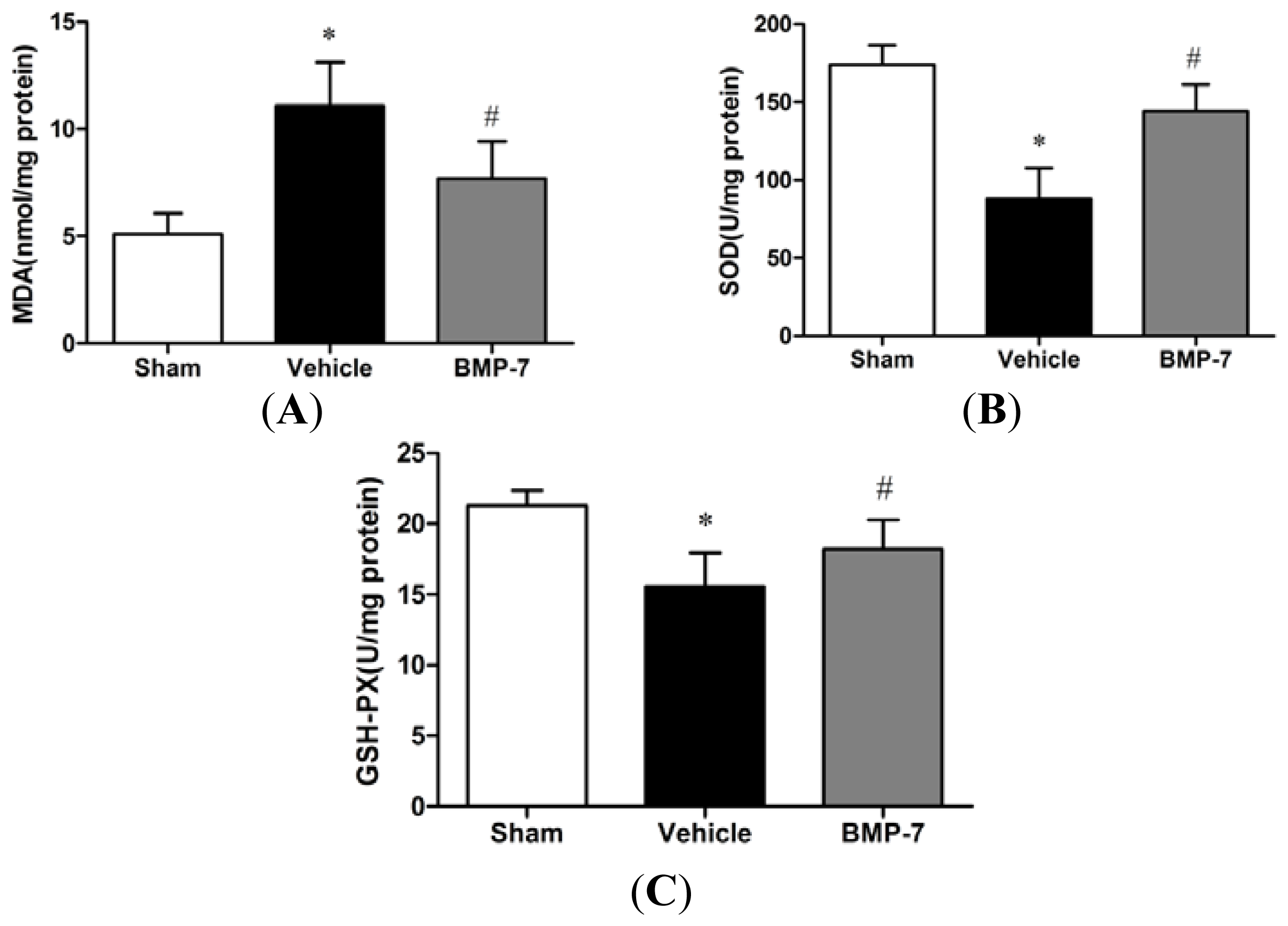
2.4. Effects of BMP-7 on MDA Content and Antioxidant Enzymes Activities
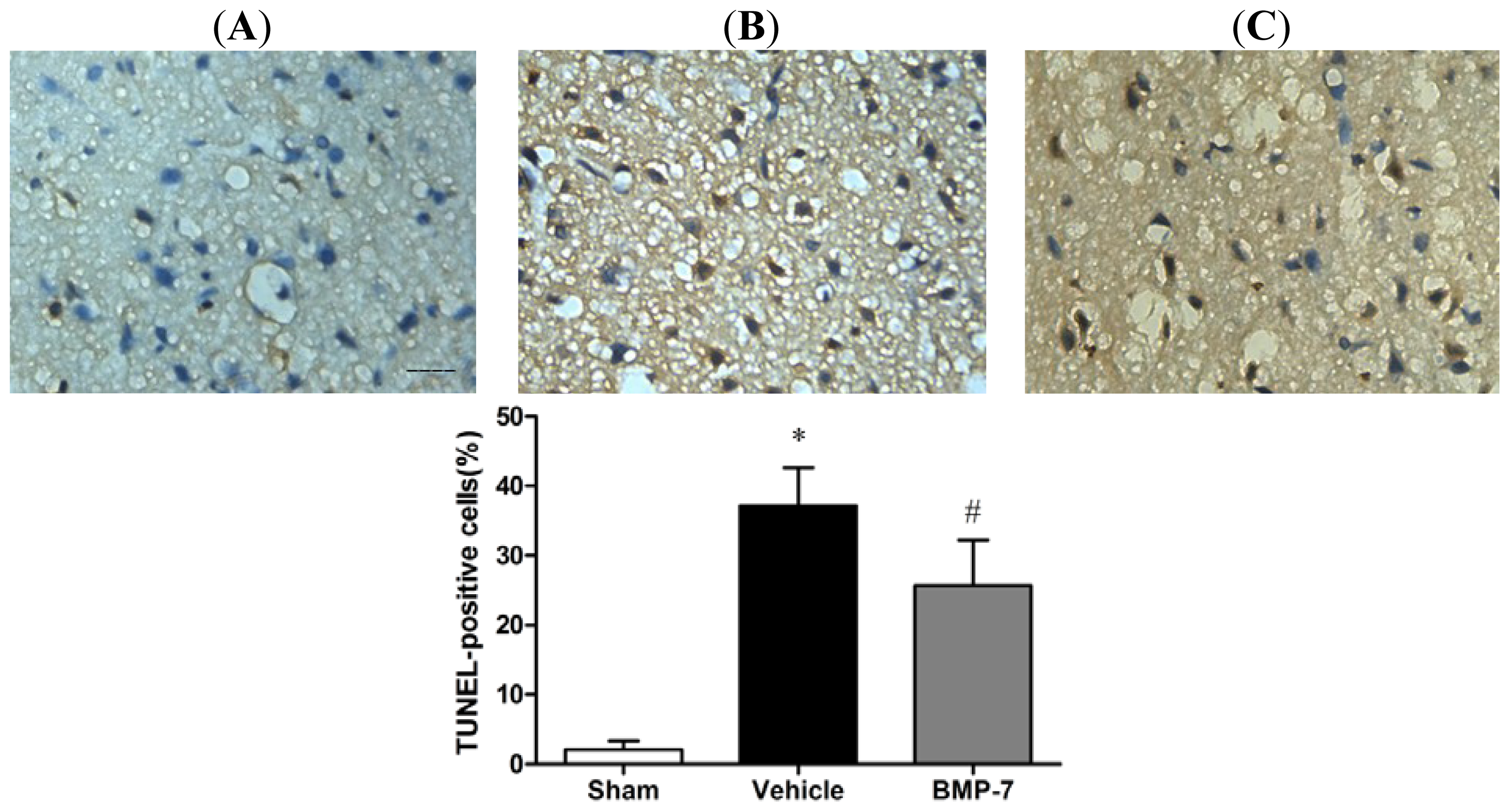
2.5. Effects of BMP-7 on Neuronal Apoptosis
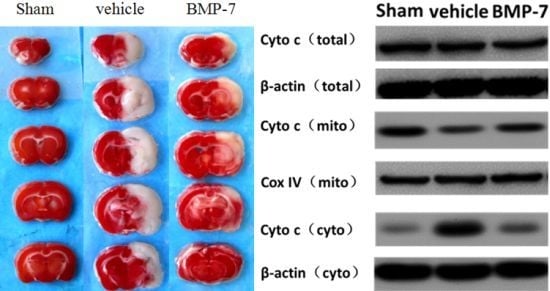
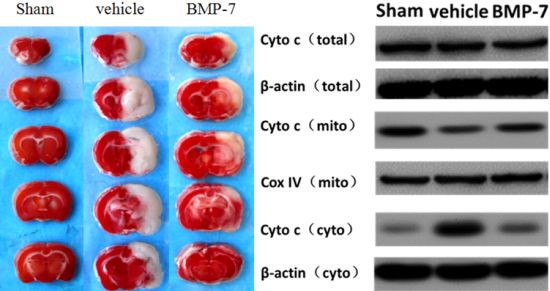
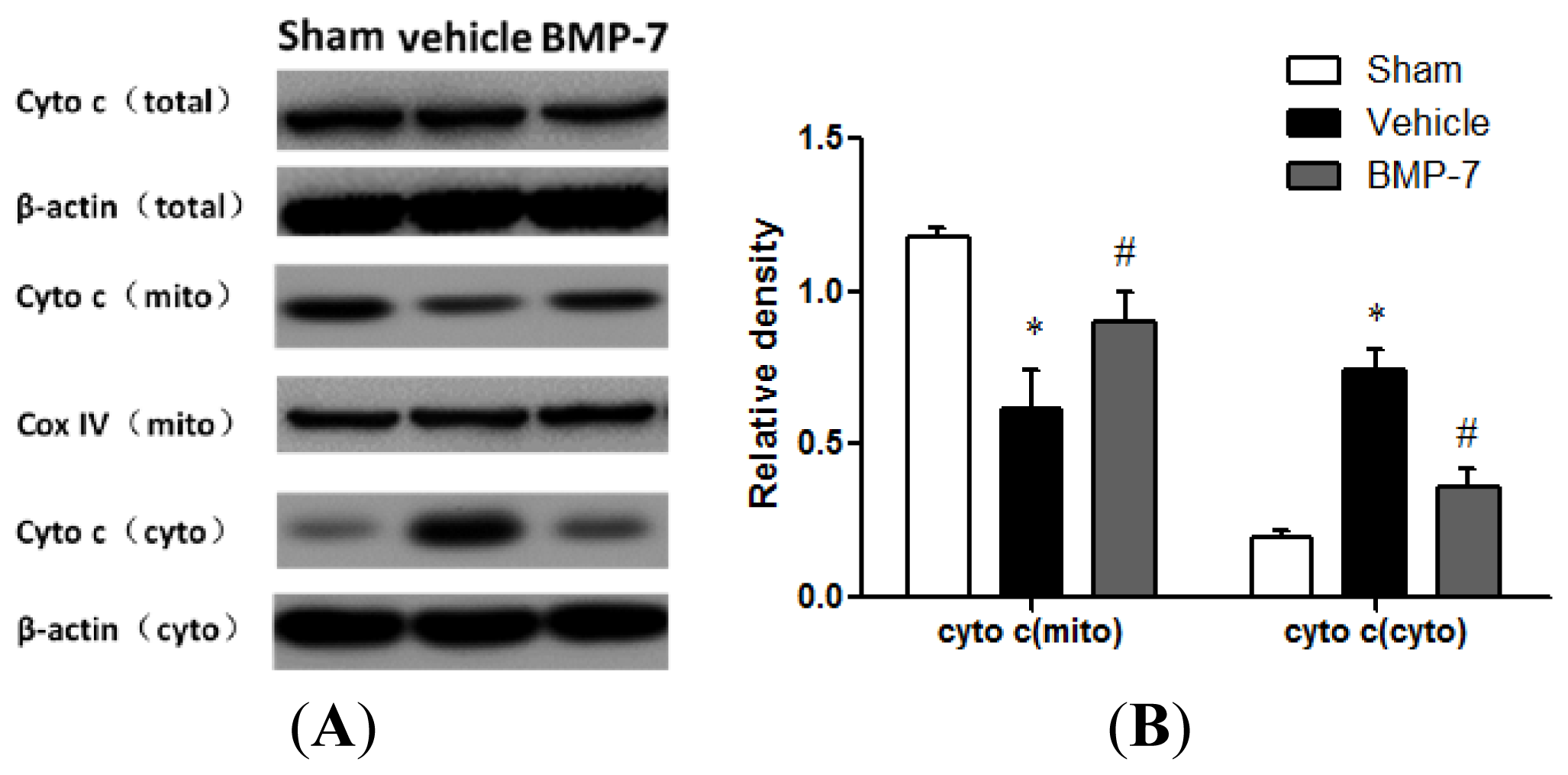
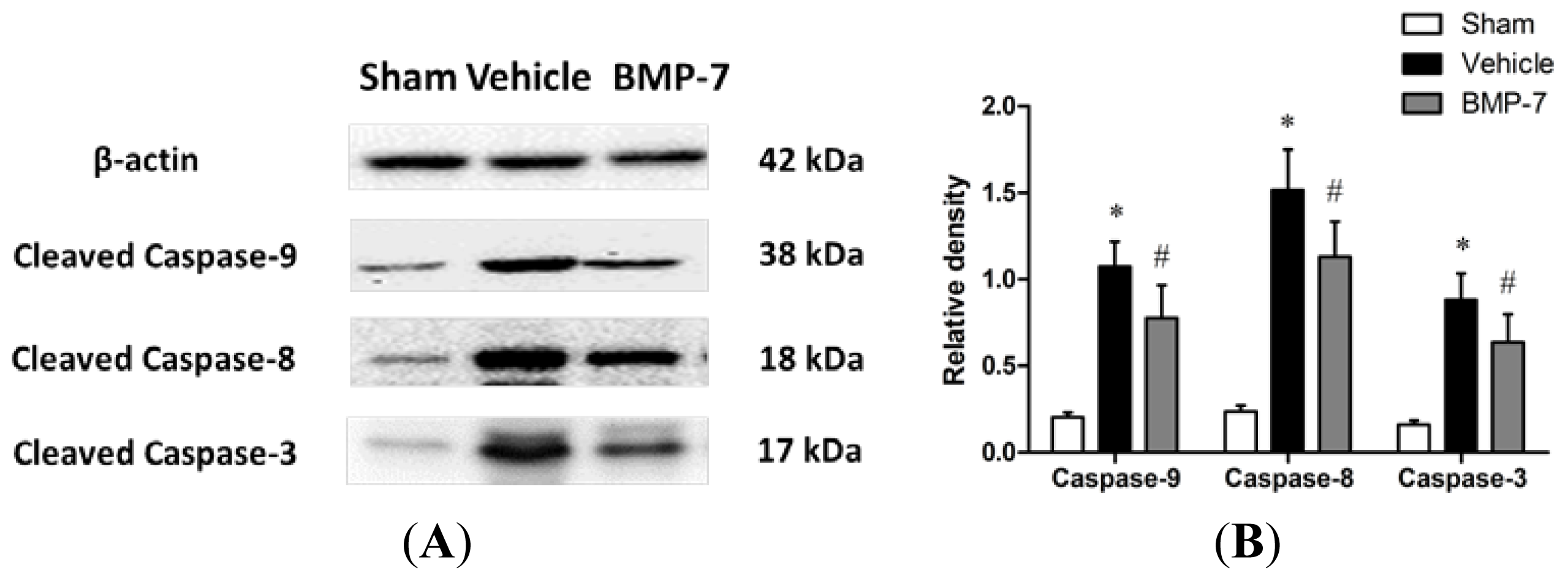
2.6. Effects of BMP-7 on Cytochrome C Release, Cleaved Caspase-3, Cleaved Caspase-9 and Cleaved Caspase-8 Expression after Cerebral IR Injury
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Groups
4.3. Induction of Cerebral IR Injury
4.4. Neurological Evaluation
4.5. Measurement of Infarct Volume
4.6. Histological Assessment
4.7. TUNEL Staining
4.8. SOD, GSH-PX Activity, and MDA Content Measurement
4.9. Preparation of Mitochondrial and Cytosolic Fraction
4.10. Western Blot Analysis
4.11. Statistical Analyses
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Flynn, R.W.; MacWalter, R.S.; Doney, A.S. The cost of cerebral ischaemia. Neuropharmacology 2008, 55, 250–256. [Google Scholar]
- Okoro, C.; Balluz, L.; Campbell, V.A.; Holt, J.B.; Mokdad, A.H. State and metropolitan-area estimates of disability in the United States, 2001. Am. J. Public Health 2005, 95, 1964–1969. [Google Scholar]
- Durukan, A.; Tatlisumak, T. Acute ischemia stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav 2007, 87, 179–197. [Google Scholar]
- Green, A.R.; Shuaib, A. Therapeutic strategies for the treatment of stroke. Drug Discov. Today 2006, 11, 681–693. [Google Scholar]
- Soderstrom, S.; Bengtsson, H.; Ebendal, T. Expression of serine/threonine kinase receptors including the bone morphogenetic factor type II receptor in the developing and adult rat brain. Cell Tissue Res 1996, 286, 269–279. [Google Scholar]
- Chen, H.L.; Lein, P.J.; Wang, J.Y.; Gash, D.; Hoffer, B.J.; Chiang, Y.H. Expression of bone morphogenetic proteins in the brain during normal aging and in 6-hydroxydopamine-lessioned animals. Brain Res 2003, 994, 81–90. [Google Scholar]
- Charytoniuk, D.A.; Traiffort, E.; Pinard, E.; Issertial, O.; Seylaz, J.; Ruat, M. Distribution of bone morphogenetic protein and bone morphogenetic protein receptor transcripts in the rodent nervous system and up-regulation of bone morphogenetic protein receptor type II in hippocampal dentate gyrus in a rat model of global cerebral ischemia. Neuroscience 2000, 100, 33–43. [Google Scholar]
- Chang, C.F.; Lin, S.Z.; Chiang, Y.H.; Morales, M.; Chou, J.; Lei, P.; Chen, H.L.; Hoffer, B.J.; Wang, Y. Intravenous administration of bone morphogenetic protein-7 after ischemia improves motor function in stroke rats. Stroke 2003, 34, 558–564. [Google Scholar]
- Shen, H.; Luo, Y.; Kuo, C.C.; Wang, Y. BMP7 reduced synergistic injury induced by methamphetamine and ischemia in mouse brain. Neurosci. Lett 2008, 442, 15–18. [Google Scholar]
- Perides, G.; Jensen, F.E.; Edgecomb, P.; Rueger, D.C.; Charness, M.E. Neuroprotective effect of human osteogenic protein-1 in a rat model of cerebral hypoxia/ischemia. Neurosci. Lett 1995, 187, 21–24. [Google Scholar]
- Lin, S.Z.; Hoffer, B.J.; Kaplan, P.; Wang, Y. Osteogenic protein-1 protects against cerebral infarction induced by MCA-ligation in adult rats. Stroke 1999, 30, 126–133. [Google Scholar]
- Chiang, Y.H.; Borlongan, C.V.; Zhou, F.C.; Hoffer, B.J.; Wang, Y. Transplantation of fetal kidney cells: Neuroprotection and neuroregeneration. Cell Transpl 2005, 14, 1–9. [Google Scholar]
- Harvey, B.K.; Hoffer, B.J.; Wang, Y. Stroke and TGF-β proteins: Glial cell line-derived neurotrophic factor and bone morphogenetic protein. Pharmacol. Ther 2005, 105, 113–125. [Google Scholar]
- Ren, J.; Kaplan, P.L.; Charette, M.F.; Speller, H.; Finklestein, S.P. Time window of intracisternal osteogenic protein-1 in enhancing functional recovery after stroke. Neuropharmacology 2000, 39, 860–865. [Google Scholar]
- Chou, J.; Harvey, B.K.; Chang, C.F.; Hoffer, B.J.; Wang, Y. Neuroregenerative effects of BMP7 after stroke in rats. Neurol. Sci 2006, 240, 21–29. [Google Scholar]
- Cox, S.; Harvey, B.K.; Sanchez, J.F.; Wang, J.Y.; Wang, Y. Mediation of BMP7 neuroprotection by MAPK and PKC IN rat primary cortical cultures. Brain Res 2004, 1010, 55–61. [Google Scholar]
- Yabe, T.; Samuels, I.; Schwartz, J.P. Bone morphogenetic proteins BMP-6 and BMP-7 have differential effects on survival and neurite outgrowth of cerebellar granule cell neurons. J. Neurosci. Res 2002, 68, 161–168. [Google Scholar]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 45, e331–e339. [Google Scholar]
- Manzanero, S.; Santro, T.; Arumugam, T.V. Neuronal oxidative stress in acute ischemic stroke: Sources and contribution to cell injury. Neurochem. Int 2013, 62, 712–718. [Google Scholar]
- Simic, P.; Vukicevic, S. Bone morphogenetic proteins: From developmental signals to tissue regeneration. Conference on bone morphogenetic proteins. EMBO Rep 2007, 8, 327–331. [Google Scholar]
- Zhen-Qiang, F.; Bing-Wei, Y.; Yong-Liang, L.; Xiang-Wei, W.; Shan-Hong, Y.; Yuan-Ning, Z.; Wei-Sheng, J.; Wei, C.; Ye, G. Localized expression of human BMP-7 by BM-MSCs enhances renal repair in an in vivo model of ischemia-reperfusion injury. Genes Cells 2012, 17, 53–64. [Google Scholar]
- Radhakrishnan, R.S.; Radhakrishnan, G.L.; Radhakrishnan, H.R.; Xue, H.; Adams, S.D.; Moore-Olufemi, S.D.; Harting, M.T.; Cox, C.S., Jr.; Kone, B.C. Pretreatment with bone morphogenetic protein-7 (BMP-7) mimics ischemia preconditioning following intestinal ischemia/reperfusion injury in the intestine and liver. Shock 2008, 30, 532–536. [Google Scholar]
- Xu, J.H.; Zhao, Y.Y.; Wang, J.K.; Yuan, Z.G.; Zhang, T.Z. Effects of mouse recombinant bone morphogenetic protein-7 transfection on cell apoptosis, NF-kappaB, and downstream genes in cultured primary cardiomyocytes after stimulated ischemia and reperfusion injury. J. Cardiovasc. Pharmacol 2010, 56, 69–77. [Google Scholar]
- Loh, K.P.; Huang, S.H.; de Silva, R.; Tan, B.K.; Zhu, Y.Z. Oxidative stress: Apoptosis in neuronal injury. Curr. Alzheimer Res 2006, 3, 327–337. [Google Scholar]
- Taylor, J.M.; Crack, P.J. Impact of oxidative stress on neuronal survival. Clin. Exp. Pharmacol. Physiol 2004, 31, 397–406. [Google Scholar]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab 2001, 21, 2–14. [Google Scholar]
- Candelario-Jalil, E.; Mhadu, N.H.; Al-Dalain, S.M.; Martinez, G.; Leon, O.S. Time course of oxidative damage in different brain regions following transient cerebral ischemia in gerbils. Neurosci. Res 2001, 41, 233–241. [Google Scholar]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar]
- Sims, N.R.; Anderson, M.F. Mitochondrial contributions to tissue damage in stroke. Neurochem. Int 2002, 40, 511–526. [Google Scholar]
- Jordan, J.; de Groot, P.W.; Galindo, M.F. Mitochondria: The head quarters in ischemia-induced neuronal death. Cent. Nerv. Syst. Aqents Med. Chem 2011, 11, 98–106. [Google Scholar]
- Xu, J.H.; Zhang, T.Z.; Zhao, Y.Y.; Wang, J.K.; Yuan, Z.G. Protective effects of recombinant human bone morphogenetic protein-7 on focal cerebral ischemia-reperfusion injury. Int. J. Neurosci 2013, 123, 375–384. [Google Scholar]
- Guan, J.; Li, H.; Lv, T.; Chen, D.; Yuan, Y.; Qu, S. Bone morphogenetic protein-7 (BMP-7) mediates ischemic preconditioning-induced ischemic tolerance via attenuating apoptosis in rat brain. Biochem. Biophys. Res. Commun 2013. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev 2001, 15, 2922–2933. [Google Scholar]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 2010, 1802, 80–91. [Google Scholar]
- Chan, P.H. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem. Res 2004, 29, 1943–1949. [Google Scholar]
- Kuida, K. Caspase-9. Int. J. Biochem. Cell Biol 2000, 32, 121–124. [Google Scholar]
- Kadirvel, R.; Ding, Y.H.; Dai, D.; Lewis, D.A.; Cloft, H.J.; Kallmes, D.F. Molecular indices of apoptosis activation in elastase-induced aneurysms after embolization with platinum coils. Stroke 2007, 38, 2787–2794. [Google Scholar]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar]







© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pei, H.; Cao, D.; Guo, Z.; Liu, G.; Guo, Y.; Lu, C. Bone Morphogenetic Protein-7 Ameliorates Cerebral Ischemia and Reperfusion Injury via Inhibiting Oxidative Stress and Neuronal Apoptosis. Int. J. Mol. Sci. 2013, 14, 23441-23453. https://doi.org/10.3390/ijms141223441
Pei H, Cao D, Guo Z, Liu G, Guo Y, Lu C. Bone Morphogenetic Protein-7 Ameliorates Cerebral Ischemia and Reperfusion Injury via Inhibiting Oxidative Stress and Neuronal Apoptosis. International Journal of Molecular Sciences. 2013; 14(12):23441-23453. https://doi.org/10.3390/ijms141223441
Chicago/Turabian StylePei, Haitao, Dongming Cao, Zhuangli Guo, Guofang Liu, Yunliang Guo, and Chenglong Lu. 2013. "Bone Morphogenetic Protein-7 Ameliorates Cerebral Ischemia and Reperfusion Injury via Inhibiting Oxidative Stress and Neuronal Apoptosis" International Journal of Molecular Sciences 14, no. 12: 23441-23453. https://doi.org/10.3390/ijms141223441
APA StylePei, H., Cao, D., Guo, Z., Liu, G., Guo, Y., & Lu, C. (2013). Bone Morphogenetic Protein-7 Ameliorates Cerebral Ischemia and Reperfusion Injury via Inhibiting Oxidative Stress and Neuronal Apoptosis. International Journal of Molecular Sciences, 14(12), 23441-23453. https://doi.org/10.3390/ijms141223441