Cell Survival and Apoptosis Signaling as Therapeutic Target for Cancer: Marine Bioactive Compounds


Abstract
:
1. Introduction
2. Cell Survival
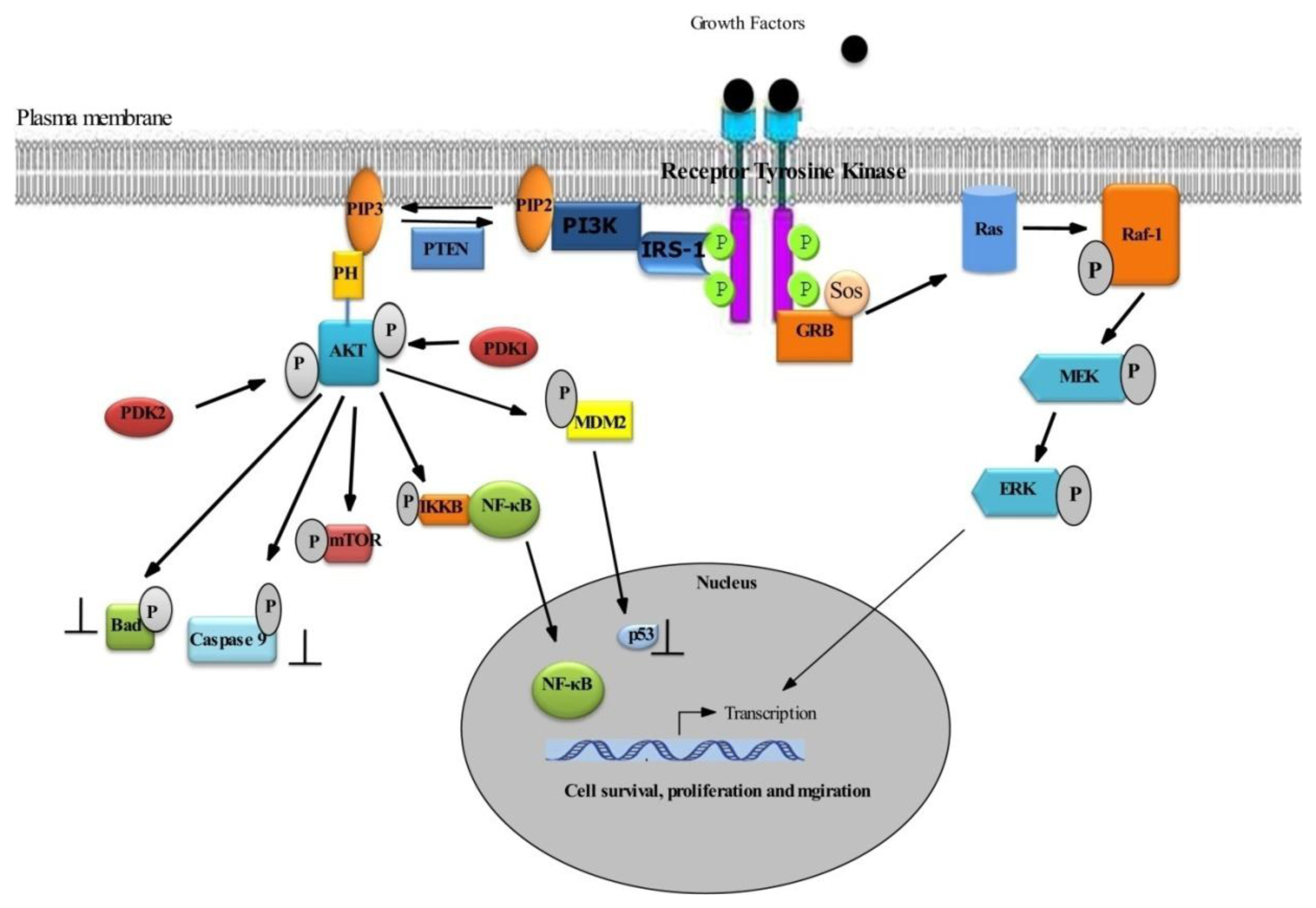
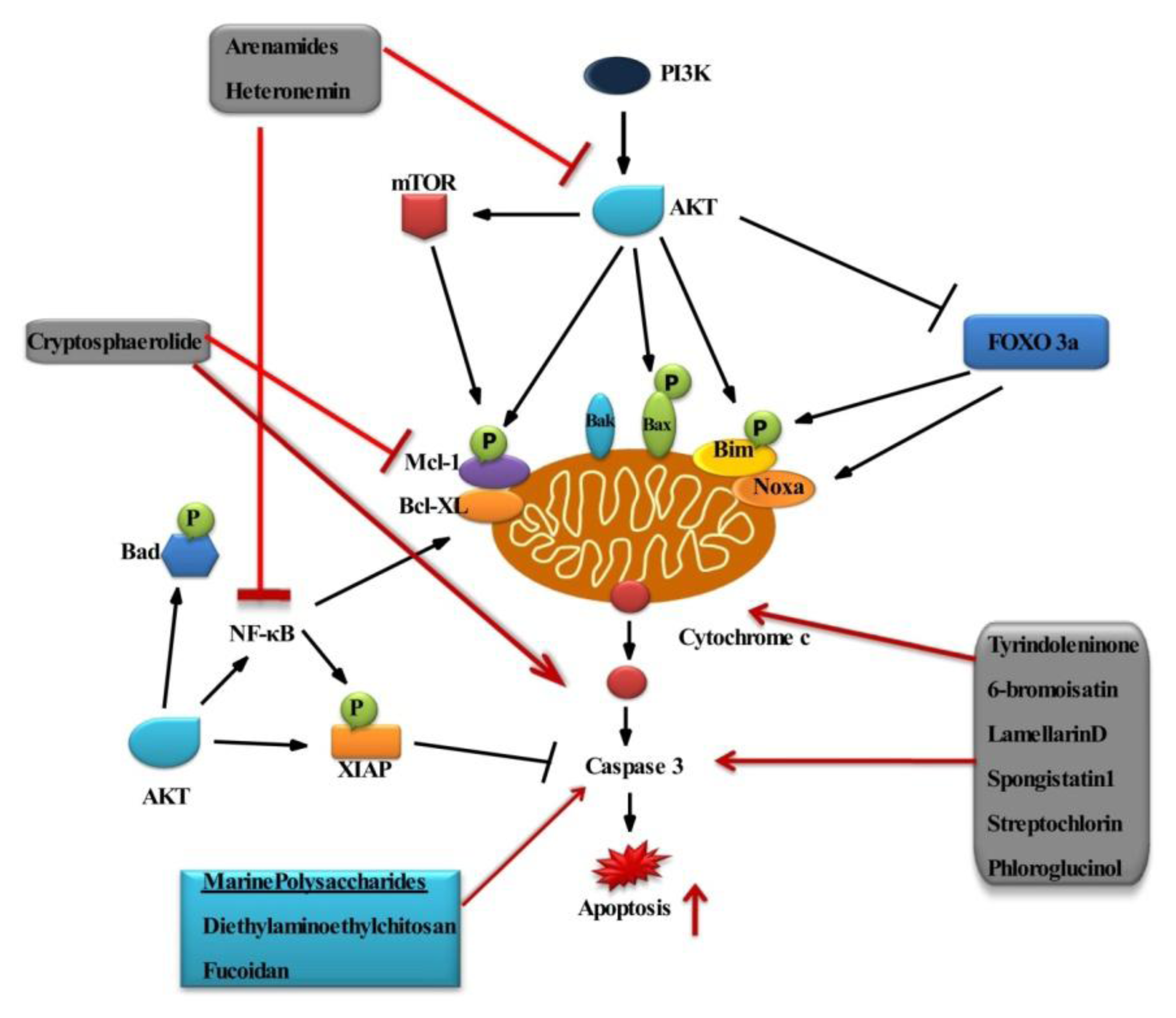
2.1. Survival Signaling via PI3K/AKT
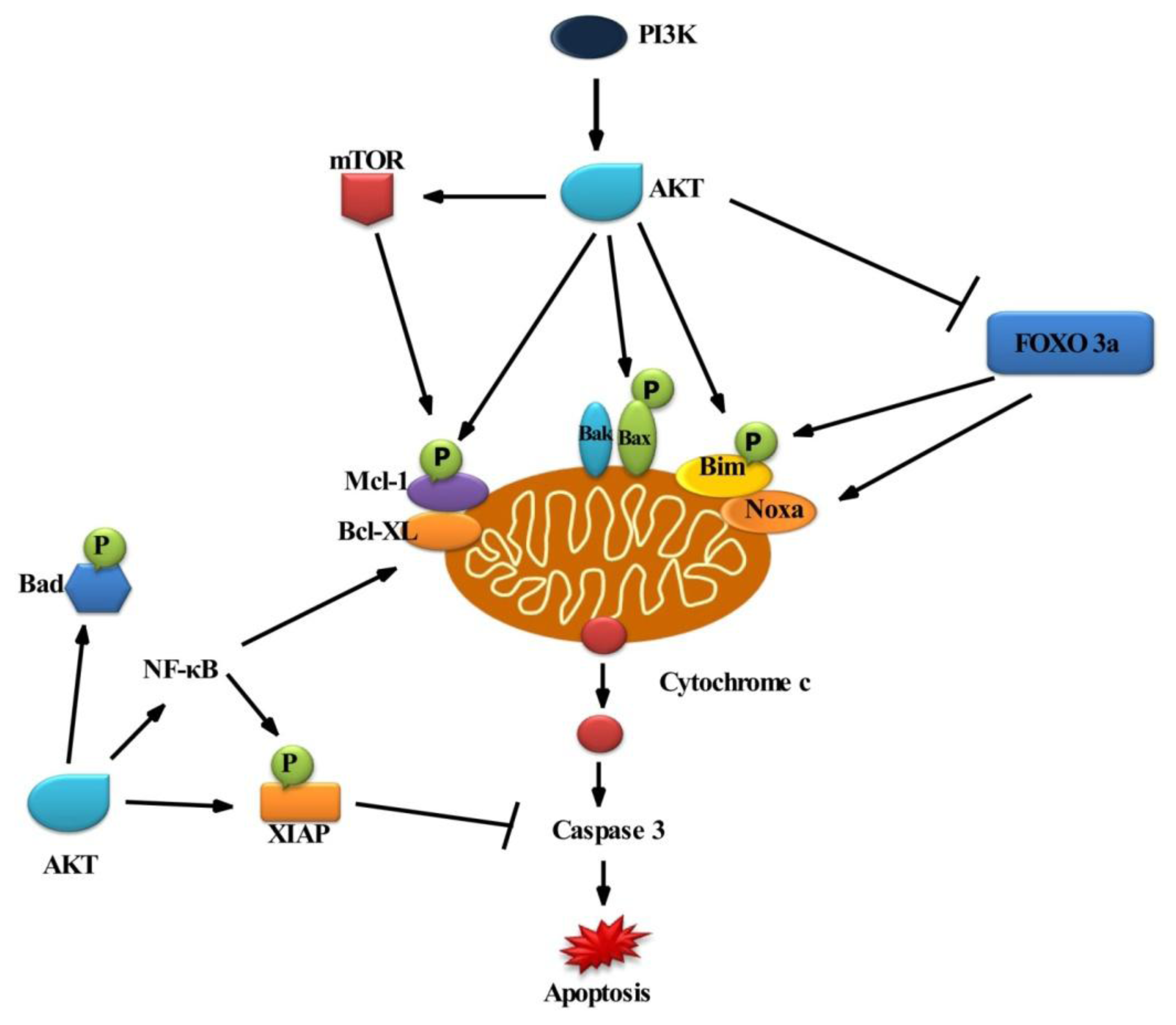
2.2. Role of AKT in Cell Survival
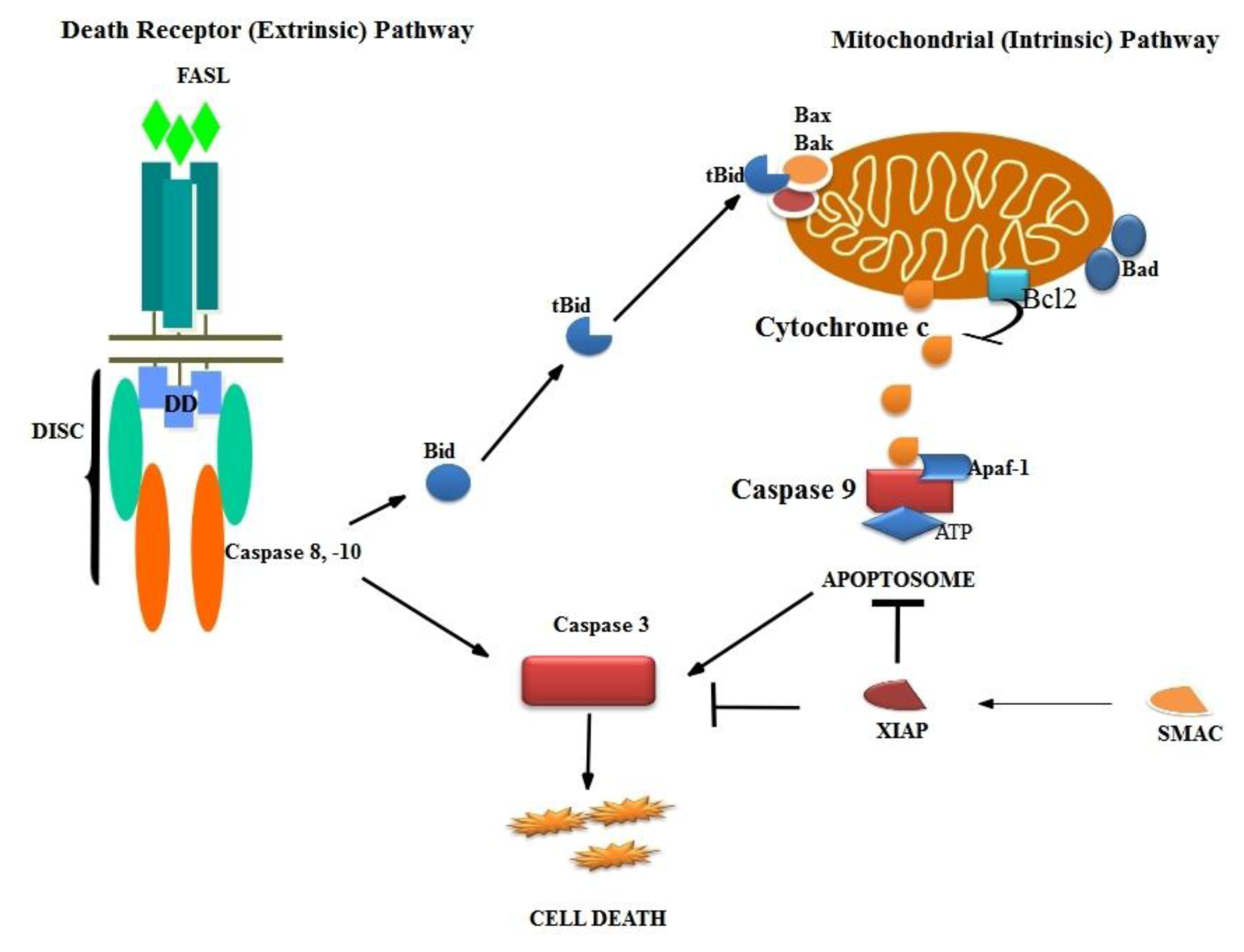
3. Apoptosis
3.1. Mechanism of Apoptosis
3.2. Intrinsic Pathway
3.3. Death Receptor Pathway
3.4. Final Pathway
3.5. Biochemical Characteristics of Apoptosis
4. Chemoprevention
The Potential Effect of Marine Bioactive Compounds on Cancer Cell Survival and Apoptosis
5. Conclusion
Acknowledgements
Conflict of Interest
References
- Lockshin, R.A.; Zakeri, Z. Cell death in health and disease. J. Cell. Mol. Med 2007, 11, 1214–1224. [Google Scholar]
- Duprez, L.; Wirawan, E.; Vanden Berghe, T.; Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect 2009, 11, 1050–1062. [Google Scholar]
- Sprick, M.R.; Walczak, H. The interplay between the Bcl-2 family and death receptor-mediated apoptosis. Biochim. Biophys. Acta 2004, 1644, 125–132. [Google Scholar]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol 2010, 72, 19–44. [Google Scholar]
- Engelman, J.A. Targeting PI3K signaling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov 2010, 9, 447–464. [Google Scholar]
- Parcellier, A.; Tintignac, L.A.; Zhuravleva, E.; Hemmings, B.A. PKB and the mitochondria: AKT signaling on apoptosis. Cell. Signal 2008, 20, 21–30. [Google Scholar]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar]
- Hsieh, A.C.; Truitt, M.L.; Ruggero, D. Oncogenic AKT activation of translation as a therapeutic target. Br. J. Cancer 2011, 105, 329–336. [Google Scholar]
- Yin, Y.; Shen, W.H. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar]
- Yamaguchi, H.; Wang, H.G. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Baxc onformational change. Oncogene 2001, 20, 7779–7786. [Google Scholar]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem 2004, 279, 21085–21095. [Google Scholar]
- Qi, X.J.; Wildey, G.M.; Howe, P.H. Evidence that Ser87 of Bim EL is phosphorylated by Akt and regulates BimEL apoptotic function. J. Biol. Chem 2006, 281, 813–823. [Google Scholar]
- Dan, H.C.; Sun, M.; Kaneko, S.; Feldman, R.I.; Nicosia, S.V.; Wang, H.G.; Tsang, B.K.; Cheng, J.Q. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J. Biol. Chem 2004, 279, 5405–5412. [Google Scholar]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar]
- Van Der Heide, L.P.; Hoekman, M.F.M.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J 2004, 380, 297–309. [Google Scholar]
- De Keizer, P.L.; Burgering, B.M.; Dansen, T.B. Forkhead boxo as a sensor, mediator and regulator of redox signaling. Antioxid. Redox Signal 2011, 14, 1093–1106. [Google Scholar]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappa B activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar]
- Bender, A.; Opel, D.; Naumann, I.; Kappler, R.; Friedman, L.; von Schweinitz, D.; Debatin, K.M.; Fulda, S. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2proteins and enhanced mitochondrial apoptosis. Oncogene 2011, 30, 494–503. [Google Scholar]
- Opel, D.; Naumann, I.; Schneider, M.; Bertele, D.; Debatin, K.M.; Fulda, S. Targeting aberrant PI3K/Akt activation by PI 103 restores sensitivity to TRAIL-induced apoptosis in neuroblastoma. Clin. Cancer Res 2011, 17, 3233–3247. [Google Scholar]
- Obexer, P.; Geiger, K.; Ambros, P.F.; Meister, B.; Ausserlechner, M.J. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ 2007, 14, 534–547. [Google Scholar]
- Ploner, C.; Kofler, R.; Villunger, A. Noxa: At the tip of the balance between life and death. Oncogene 2008, 27, 84–92. [Google Scholar]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis, a link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anti cancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov 2012, 11, 109–124. [Google Scholar]
- Levine, A. P53, the cellular gatekeeper for growth and division. Cell. Mol. Biol 1997, 88, 323–331. [Google Scholar]
- Yin, C.; Knudson, C.; Korsmeyer, S.; Dyke, T.V. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 1997, 385, 637–640. [Google Scholar]
- Pietenpol, J.; Stewart, Z. Cell cycle checkpoint signaling, cell cycle arrest versus apoptosis. Toxicology 2002, 475, 181–182. [Google Scholar]
- Johnson, T.; Yu, Z.; Ferrans, V.; Lowenstein, R.; Finkel, T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 11848–11852. [Google Scholar]
- Sheikh, M.; Fornace, A.J. Role of p53 family members in apoptosis. J. Cell. Physiol 2000, 182, 171–181. [Google Scholar]
- Martindale, J.; Holbrook, N. Cellular response to oxidative stress, signaling for suicide and survival. J. Cell. Physiol 2002, 192, 1–15. [Google Scholar]
- Saelens, X.; Festjens, N.; Walle, L.V.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar]
- Van Loo, G.; van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ 2002, 9, 20–26. [Google Scholar]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 2006, 13, 1423–1433. [Google Scholar]
- Chinnaiyan, A.M. The apoptosome, heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J 2002, 23, 2134–2145. [Google Scholar]
- Schimmer, A.D. Inhibitor of apoptosis proteins, translating basic knowledge into clinical practice. Cancer Res 2004, 64, 7183–7190. [Google Scholar]
- Ekert, P.G.; Vaux, D.L. The mitochondrial death squad, hardened killers or innocent bystanders? Curr. Opin. Cell Biol 2005, 17, 626–630. [Google Scholar]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.; Sasaki, T.; Elia, A.J.; Cheng, H.Y.M.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prévost, M.-C.; et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med 2000, 192, 571–580. [Google Scholar]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar]
- Cory, S.; Adams, J.M. The Bcl2 family, regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar]
- Schuler, M.; Green, D.R. Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans 2001, 29, 684–688. [Google Scholar]
- Vaux, D.L.; Cory, S.; Adamsm, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar]
- Weyhenmeyer, B.; Murphy, A.C.; Prehn, J.H.; Murphy, B.M. Targeting the anti-apoptotic bcl-2 family members for the treatment of cancer. Exp. Oncol 2012, 34, 192–199. [Google Scholar]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t (14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar]
- Reed, J.C. Bcl-2 family proteins, regulators of apoptosis and chemoresistance in hematologic malignancies. Semin. Hematol 1997, 34, 9–19. [Google Scholar]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, T.M. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar]
- Reed, J.C. Bcl-2 and the regulation of programmed cell death. J. Cell. Biol 1994, 124, 1–6. [Google Scholar]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins amd cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar]
- Scorrano, L.; Korsmeyer, S. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem. Biophys. Res. Commun 2003, 304, 437–444. [Google Scholar]
- Korsmeyer, S.J. Regulators of cell death. Trends Genet 1995, 11, 101–105. [Google Scholar]
- Lee, H.J.; Lee, H.J.; Lee, E.O.; Ko, S.G.; Bae, H.S.; Kim, C.H.; Ahn, K.S.; Lu, J.; Kim, S.H. Mitochondria-cytochrome C-caspase-9 cascade mediates isorhamnetin-induced apoptosis. Cancer Lett 2008, 207, 342–353. [Google Scholar]
- Fiandalo, M.V.; Kyprianou, N. Caspase control: Protagonists of cancer cell apoptosis. Exp. Oncol 2012, 34, 165–175. [Google Scholar]
- Reed, J. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 9–17. [Google Scholar]
- Zapata, J.M.; Pawlowski, K.; Haas, E.; Ware, C.F.; Godzik, A.; Reed, J.C. A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J. Biol. Chem 2001, 276, 24242–24252. [Google Scholar]
- Lamy, T.; Liu, J.H.; Landowski, T.H.; Dalton, W.S.; Loughran, T.P., Jr. Dysregulation of CD95/CD95 ligand-apoptotic pathway in CD3 large granular lymphocyte leukemia. Blood 1998, 92, 4771–4777. [Google Scholar]
- Landowski, T.H.; Moscinski, L.; Burke, R.; Buyuksal, I.; Painter, J.S.; Goldstein, S.; Dalton, W.S. CD95 antigen mutations in hematopoietic malignancies. Leuk. Lymphoma 2001, 42, 835–846. [Google Scholar]
- Krammer, P.H. CD95’s deadly mission in the immune system. Nature 2000, 407, 789–795. [Google Scholar]
- Wajant, H. The Fas signaling pathway, more than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar]
- Landowski, T.H.; Qu, N.; Buyuksal, I.; Painter, J.S.; Dalton, W.S. Mutations in the Fas antigen in patients with multiple myeloma. Blood 1997, 90, 4266–4270. [Google Scholar]
- Irie, S.; Li, Y.; Kanki, H.; Ohyama, T.; Deaven, L.L.; Somlo, S.; Sato, T.A. Identification of two Fas-associated phosphatase-1 (FAP-1) promoters in human cancer cells. DNA Seq 2001, 11, 519–526. [Google Scholar]
- Krueger, A.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. FLICE-inhibitory proteins, regulators of death receptor-mediated apoptosis. Mol. Cell. Biol 2001, 21, 8247–8254. [Google Scholar]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem 2000, 69, 217–245. [Google Scholar]
- Henson, P.M.; Bratton, D.L.; Fadok, V.A. Apoptotic cell removal. Curr. Biol 2001, 11, 795–805. [Google Scholar]
- Green, D.R.; Evan, G.I. A matter of life and death. Cancer Cell 2002, 1, 19–30. [Google Scholar]
- Thornberry, N.A.; Lazebnik, Y. Caspases, enemies within. Science 1998, 281, 1312–1316. [Google Scholar]
- Mancini, M.; Nicholson, D.W.; Roy, S.; Thornberry, N.A.; Peterson, E.P.; Casciola-Rosen, L.A.; Rosen, A. The caspase-3 precursor has a cytosolic and mitochondrial distribution, implications for apoptotic signaling. J. Cell Biol 1998, 140, 1485–1495. [Google Scholar]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar]
- Cohen, G.M. Caspases, the executioners of apoptosis. Biochem. J 1997, 336, 1–16. [Google Scholar]
- Rai, N.K.; Tripathi, K.; Sharma, D.; Shukla, V.K. Apoptosis, a basic physiologic process in wound healing. Int. J. Low Extrem. Wounds 2005, 4, 138–144. [Google Scholar]
- Hu, S.; Snipas, S.J.; Vincenz, C.; Salvesen, G.; Dixit, V.M. Caspase-14 is a novel developmentally regulated protease. J. Biol. Chem 1998, 273, 29648–29653. [Google Scholar]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar]
- Koenig, U.; Eckhart, L.; Tschachler, E. Evidence that caspase-13 is not a human but a bovine gene. Biochem. Biophys. Res. Commun 2001, 285, 1150–1154. [Google Scholar]
- Kang, S.J.; Wang, S.; Kuida, K.; Yuan, J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ 2002, 9, 1115–1125. [Google Scholar]
- Nemes, Z., Jr; Friis, R.R.; Aeschlimann, D.; Saurer, S.; Paulsson, M.; Fésüs, L. Expression and activation of tissue transglutaminase in apoptotic cells of involuting rodent mammary tissue. Eur. J. Cell Biol. 1996, 70, 125–133. [Google Scholar]
- Bortner, C.D.; Oldenburg, N.B.; Cidlowski, J.A. The role of DNA fragmentation in apoptosis. Trends Cell Biol 1995, 5, 21–26. [Google Scholar]
- Bratton, D.L.; Fadok, V.A.; Richter, D.A.; Kailey, J.M.; Guthrie, L.A.; Henson, P.M. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J. Biol. Chem 1997, 272, 26159–26165. [Google Scholar]
- Arur, S.; Uche, U.E.; Rezaul, K.; Fong, M.; Scranton, V.; Cowan, A.E.; Mohler, W.; Han, D.K. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev. Cell 2003, 4, 587–598. [Google Scholar]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar]
- Kuno, T.; Tsukamoto, T.; Hara, A.; Tanaka, T. Cancer chemoprevention through the induction of apoptosis by natural compounds. J. Biophys. Chem 2012, 3, 156–173. [Google Scholar]
- Fisher, B.; Perera, F.; Cooke, A.; Opeitum, A; Stitt, L. Long-term follow-up of axillary node-positive breast cancer patients receiving adjuvant tamoxifen alone, patterns of recurrence. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 117–123. [Google Scholar]
- Cummings, S.; Eckert, S.; Krueger, K.; Grady, D.; Powles, T.J.; Cauley, J.A.; Norton, L.; Nickelsen, T.; Bjarnason, N.H.; Morrow, M.; et al. The effect of raloxifene on risk of breast cancer in postmenopausal women, results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. J. Am. Med. Assoc 1999, 281, 2189–2197. [Google Scholar]
- Veronesi, U.; de Palo, G.; Marubini, E.; Costa, A.; Formelli, F.; Mariani, L.; Decensi, A.; Camerini, T.; del Turco, M.R.; di Mauro, M.G.; et al. Randomized trial of fenretinide to prevent second breast malignancy in women with early breast cancer. J. Natl. Cancer Inst 1999, 91, 1847–1856. [Google Scholar]
- Hail, N., Jr; Cortes, M.; Drake, E.N.; Spallholz, J.E. Cancer chemoprevention: A radical perspective. Free Rad. Biol. Med. 2008, 45, 97–110. [Google Scholar]
- Kelloff, G.J.; Sigman, C.C.; Greenwald, P. Cancer chemoprevention: Progress and promise. Eur. J. Cancer 1999, 35, 2031–2038. [Google Scholar]
- Kakizoe, T. Chemoprevention of cancer-Focusing on clinical trials. Jpn. J. Clin. Oncol 2003, 33, 421–442. [Google Scholar]
- Park, E.J.; Pezzuto, J.M. Botanicals in cancer chemoprevention. Cancer Metast. Rev 2002, 21, 231–255. [Google Scholar]
- Stan, S.D.; Kar, S.; Stoner, G.D.; Singh, S.V. Bioactive food components and cancer risk reduction. J. Cell. Biochem 2008, 104, 339–356. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod 2007, 70, 461–477. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep 2011, 28, 196–268. [Google Scholar]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug. Discov 2010, 17, 790–803. [Google Scholar]
- Gullett, N.P.; Ruhul Amin, A.R.; Bayraktar, S.; Pezzuto, J.M.; Shin, D.M.; Khuri, F.R.; Aggarwal, B.B.; Surh, Y.J.; Kucuk, O. Cancer prevention with natural compounds. Semin. Oncol 2010, 37, 258–281. [Google Scholar]
- Hickford, S.J.; Blunt, J.W.; Munro, M.H. Antitumour polyether macrolides, four new halichondrins from the New Zealand deep-water marine sponge Lissodendoryx sp. Bioorg. Med. Chem 2009, 17, 2199–2203. [Google Scholar]
- Aoki, S.; Cao, L.; Matsui, K.; Rachmat, R.; Akiyamac, S.; Kobayashia, M. Kendarimide A, a novel peptide reversing P-glycoprotein-mediated multidrug resistance in tumor cells, from a marine sponge of Haliclona sp. Tetrahedron 2004, 60, 7053–7059. [Google Scholar]
- Thomas, T.R.A.; Kavlekar, D.P.; LokaBharathi, P.A. Marine Drugs from Sponge-Microbe Association—A Review. Mar. Drugs 2010, 8, 1417–1468. [Google Scholar]
- Rodriguez, A.D.; Martinez, N. Marine antitumor agents, 14-deoxycrassin and pseudoplexaurol, new cembranoid diterpenes from the Caribbean gorgonian Pseudoplexaura porosa. Experientia 1993, 49, 179–181. [Google Scholar]
- Olano, C.; Méndez, C.; Salas, J.A. Antitumor Compounds from Marine Actinomycetes. Mar. Drugs 2009, 7, 210–248. [Google Scholar]
- Hassan, H.M.; Khanfar, M.A.; Elnagar, A.Y.; Mohammed, R.; Shaala, L.A.; Youssef, D.T.A.; Hifnawy, M.S.; Sayed, K.A.E. Pachycladins A–E, prostate cancer invasion and migration inhibitory Eunicellin-based diterpenoids from the red sea soft coral Cladiella pachyclados. J. Nat. Prod 2010, 73, 848–853. [Google Scholar]
- Luqman, S.; Pezzuto, J.M. NFkappa B: A promising target for natural products in cancer chemoprevention. Phytother. Res 2010, 24, 949–963. [Google Scholar]
- Karin, M. Nuclear factor-kappa B in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar]
- Crowell, J.A.; Steele, V.E.; Fay, J.R. Targeting the AKT protein kinase for cancer chemoprevention. Mol. Cancer Therap 2007, 6, 2139–2148. [Google Scholar]
- Andersen, J.N.; Sathyanarayanan, S.; di Bacco, A.; Chi, A.; Zhang, T.; Chen, A.H.; Dolinski, B.; Kraus, M.; Roberts, B.; Arthur, W.; et al. Pathway-based identification of biomarkers for targeted therapeutics, Personalized oncology with PI3K pathway inhibitors. Sci. Transl. Med 2010, 2, 43–55. [Google Scholar]
- Asolkar, R.N.; Freel, K.C.; Jensen, P.R.; Fenical, W.; Kondratyuk, T.P.; Park, E.J.; Pezzuto, J.M. Arenamides A–C, cytotoxic NF kappa B inhibitors from the marine actinomycetes Salinispora arenicola. J. Nat. Prod 2009, 72, 396–402. [Google Scholar]
- Schumacher, M.; Cerella, C.; Eifes, S.; Chateauvieux, S.; Morceau, F.; Jaspars, M.; Dicato, M.; Diederich, M. Heteronemin, a spongean sesterterpene, inhibits TNF alpha-induced NF-kappa B activation through proteasome inhibition and induces apoptotic cell death. Biochem. Pharmacol 2010, 79, 610–622. [Google Scholar]
- Edwards, V.; Benkendorff, K.; Young, F. Marine Compounds Selectively Induce Apoptosis in Female Reproductive Cancer Cells but Not in Primary-Derived Human Reproductive Granulosa Cells. Mar. Drugs 2012, 10, 64–83. [Google Scholar]
- Oh, H.; Jensen, P.R.; Murphy, B.T.; Fiorilla, C.; Sullivan, J.F.; Ramsey, T.; Fenical, W. Cryptosphaerolide, a cytotoxic Mcl-1 inhibitor from a marine-derived ascomycete related to the genus Cryptosphaeria. J. Nat. Prod 2010, 73, 998–1001. [Google Scholar]
- Dias, N.; Vezin, H.; Lansiaux, A.; Bailly, C. Topoisomerase Inhibitors of Marine Origin and Their Potential Use as Anticancer Agents. Top. Curr. Chem 2005, 253, 89–108. [Google Scholar]
- Bonnard, I.; Bontemps, N.; Lahmy, S.; Banaigs, B.; Combaut, G.; Francisco, C.; Colson, P.; Houssier, C.; Waring, M.J.; Bailly, C. Binding to DNA and cytotoxic evaluation of ascididemin, the major alkaloid from the Mediterranean ascidian Cystodytes dellechiajei. Anticancer Drug Des 1995, 10, 333–346. [Google Scholar]
- Dassonneville, L.; Wattez, N.; Baldeyrou, B.; Mahieu, C.; Lansiaux, A.; Banaigs, B.; Bonnard, I.; Bailly, C. Inhibition of topoisomerase II by the marine alkaloid ascididemin and induction of apoptosis in leukemia cells. Biochem. Pharmacol 2000, 60, 527–537. [Google Scholar]
- Tardy, C.; Facompré, M.; Laine, W.; Baldeyroua, B.; arc a-Gravalosb, D.; Franceschb, A.; Mateob, C.; Pastorb, A.; Jiménezb, J.A.; Manzanares, I.; et al. Topoisomerase I-mediated DNA cleavage as a guide to the development of antitumor agents derived from the marine alkaloid lamellarin D, triester derivatives incorporating amino acid residues. Bioorg. Med. Chem 2004, 12, 1697–1712. [Google Scholar]
- Ballot, C.; Kluza, J.; Martoriati, A.; Nyman, U.; Formstecher, P.; Joseph, B.; Bailly, C.; Marchetti, P. Essential role of mitochondria in apoptosis of cancer cells induced by the marine alkaloid Lamellarin D. Mol. Cancer Ther 2009, 8, 3307–3317. [Google Scholar]
- Schyschka, L.; Rudy, A.; Jeremias, I.; Barth, N.; Pettit, G.R.; Vollmar, A.M. Spongistatin 1: A new chemosensitizingmarine compoundthat degrades XIAP. Leukemia 2008, 22, 1737–1745. [Google Scholar]
- Shin, H.J.; Jeong, H.S.; Lee, H.S.; Park, S.K.; Hwan, M.K.; Ho, J.K. Isolation and structure determination of streptochlorin, an antiproliferative agent from a marine-derived Streptomyces sp. 04DH110. J. Microbiol. Biotech 2007, 17, 1403–1406. [Google Scholar]
- Choi, I.K.; Shin, H.J.; Lee, H.S.; Kwon, H.J. Streptochlorin, a marine natural product, inhibits NF-κB activation and suppresses angiogenesis in vitro. J. Microbiol. Biotech 2007, 17, 1338–1343. [Google Scholar]
- Park, C.; Shin, H.J.; Kim, G.Y.; Kwon, T.K.; Nam, T.J.; Kim, S.K.; Cheong, J.; Choi, I.W.; Choi, Y.H. Induction of apoptosis by streptochlorin isolated from Streptomyces sp. in human leukemic U937 cells. Toxicol. In Vitro 2008, 22, 1573–1581. [Google Scholar]
- Ali, M.S.; Jahangir, M.; Saleem, M.; Pervez, M.K.; Hameed, S.; Ahmad, V.U. Metabolites of marine algae collected from Karachi-coasts of Arabian sea. Nat. Prod. Sci 2000, 6, 61–65. [Google Scholar]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Antioxidant properties of phenolic compounds. Trends Plant Sci 1997, 2, 152–159. [Google Scholar]
- Maegawa, M.; Yokohama, Y.; Aruga, Y. Critical light conditions for young Ecklonia cava and Eisenia bicyclis with reference to photosynthesis. Hydrobiologia 1987, 151, 447–455. [Google Scholar]
- Kong, S.C.; Kim, A.J.; Yoon, Y.N.; Kim, S.K. Induction of apoptosis by phloroglucinol derivative from Ecklonia Cava in MCF-7 human breast cancer cells. Food Chem. Toxicol 2009, 47, 1653–1658. [Google Scholar]
- Qin, C.; Du, Y.; Xiao, L.; Li, Z.; Gao, X. Enzymic preparation of water-soluble chitosan and their antitumor activity. Int. J. Biol. Macromol 2002, 31, 111–117. [Google Scholar]
- Lee, H.S.; Ryua, B.; Je, Y.J.; Kim, S.K. Diethylaminoethyl chitosan induces apoptosis in HeLa cells via activation of caspase-3 and p53 expression. Carbohydr. Polym 2011, 84, 571–578. [Google Scholar]
- Yamasaki-Miyamoto, Y.; Yamasaki, M.; Tachibana, H.; Yamada, K. Fucoidan Induces Apoptosis through Activation of Caspase-8 on Human Breast Cancer MCF-7 Cells. J. Agric. Food Chem 2009, 57, 8677–8682. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Name of the Compound | Source of Organisms | Chemical class | Cancer Target | Reference |
|---|---|---|---|---|---|
| 1 | Arenamides A–C | Actinomycete (Salinispora arenicola) | Cyclohexa-depsipeptides | Human colon carcinoma cell line (HCT-116) | [107] |
| 2 | Heteronemin | Sponge (Hyrtios sp.) | Sesterterpene | Leukemia (K562 cells) | [108] |
| 3 | 6-bromoisatin | Whelk (Dicathais orbita) | Indole derivative | Ovary, granulosa, Choriocarcinoma (OVCAR-3, KGN, Jar) | [109] |
| 4 | Tyrindoleninone | Whelk (Dicathais orbita) | Indole derivative | Ovary, granulosa, Choriocarcinoma (OVCAR-3, KGN, Jar) | [109] |
| 5 | Cryptosphaerolide | Ascomycete fungal strain CNL-523 (Cryptosphaeria sp.) | sesquiterpenoid | Human colon carcinoma cell line (HCT-116) | [110] |
| 6 | Makaluvamine A | sponge (Zyzzya fuliginosa) | pyrroloquinoline | Colon cancer (HCT-116 cells) | [111] |
| 7 | Ascididemin | Actinomycete (Salinispora arenicola) | Cyclohexa-depsipeptides | Human colon carcinoma cell line (HCT-116) | [112,113] |
| 8 | Lamellarin D | Prosobranch mollusc of the genus (Lamellaria) | Alkaloid | Leukemia | [114,115] |
| 9 | Spongistatin 1 | Sponges (Spirastrella spinispirulifera and Hyrtios erecta) | macrocyclic lactone | Leukemia (Jurkat cells) | [116] |
| 10 | Streptochlorin | Streptomyces sp. | Methyl pyridine | Leukemia (U937 cells) | [117–119] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kalimuthu, S.; Se-Kwon, K. Cell Survival and Apoptosis Signaling as Therapeutic Target for Cancer: Marine Bioactive Compounds. Int. J. Mol. Sci. 2013, 14, 2334-2354. https://doi.org/10.3390/ijms14022334
Kalimuthu S, Se-Kwon K. Cell Survival and Apoptosis Signaling as Therapeutic Target for Cancer: Marine Bioactive Compounds. International Journal of Molecular Sciences. 2013; 14(2):2334-2354. https://doi.org/10.3390/ijms14022334
Chicago/Turabian StyleKalimuthu, Senthilkumar, and Kim Se-Kwon. 2013. "Cell Survival and Apoptosis Signaling as Therapeutic Target for Cancer: Marine Bioactive Compounds" International Journal of Molecular Sciences 14, no. 2: 2334-2354. https://doi.org/10.3390/ijms14022334
APA StyleKalimuthu, S., & Se-Kwon, K. (2013). Cell Survival and Apoptosis Signaling as Therapeutic Target for Cancer: Marine Bioactive Compounds. International Journal of Molecular Sciences, 14(2), 2334-2354. https://doi.org/10.3390/ijms14022334