Effects of Heme Oxygenase-1 Upregulation on Blood Pressure and Cardiac Function in an Animal Model of Hypertensive Myocardial Infarction

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
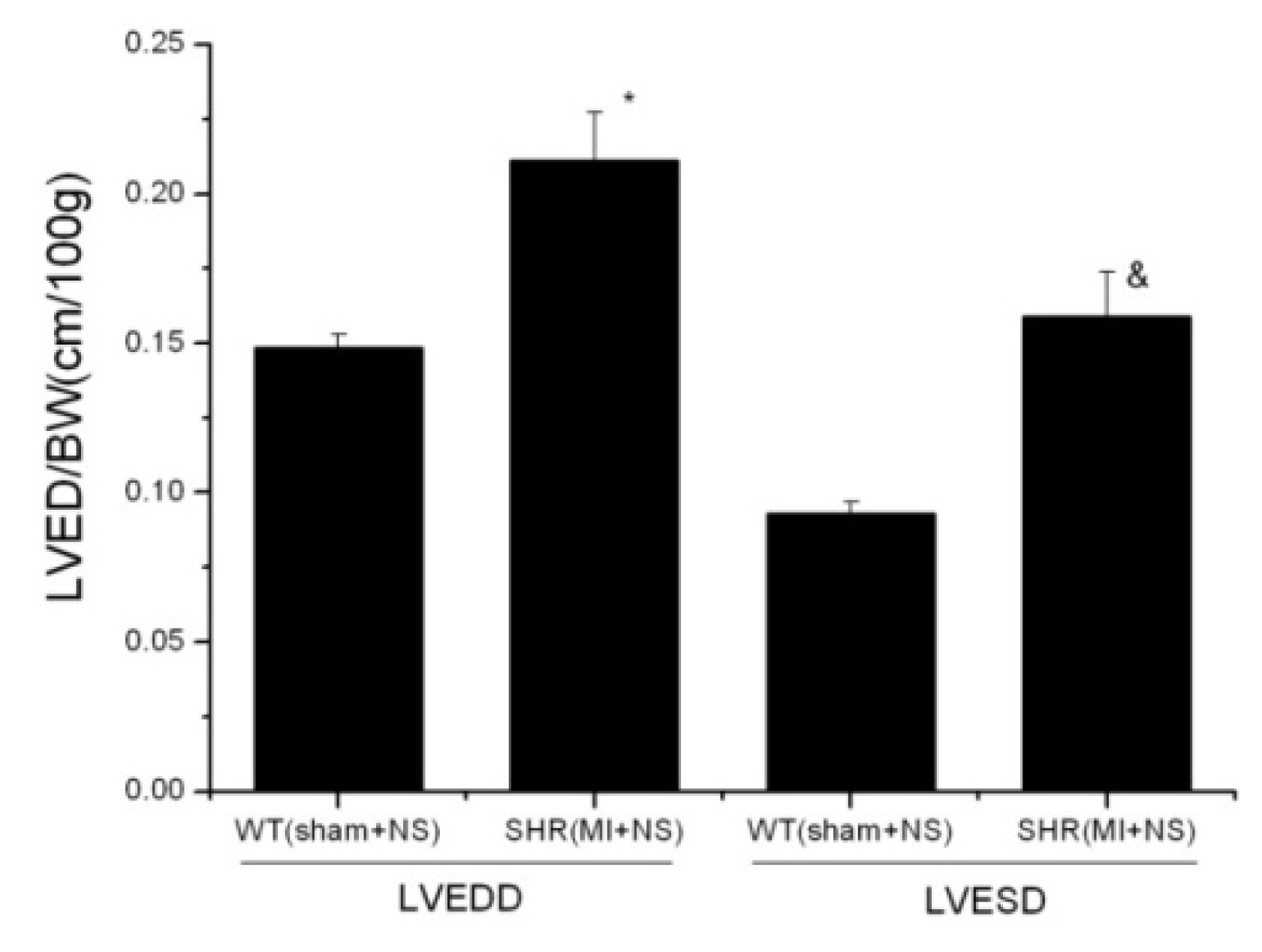
2.1.1. Baseline Characters between WT and SHR
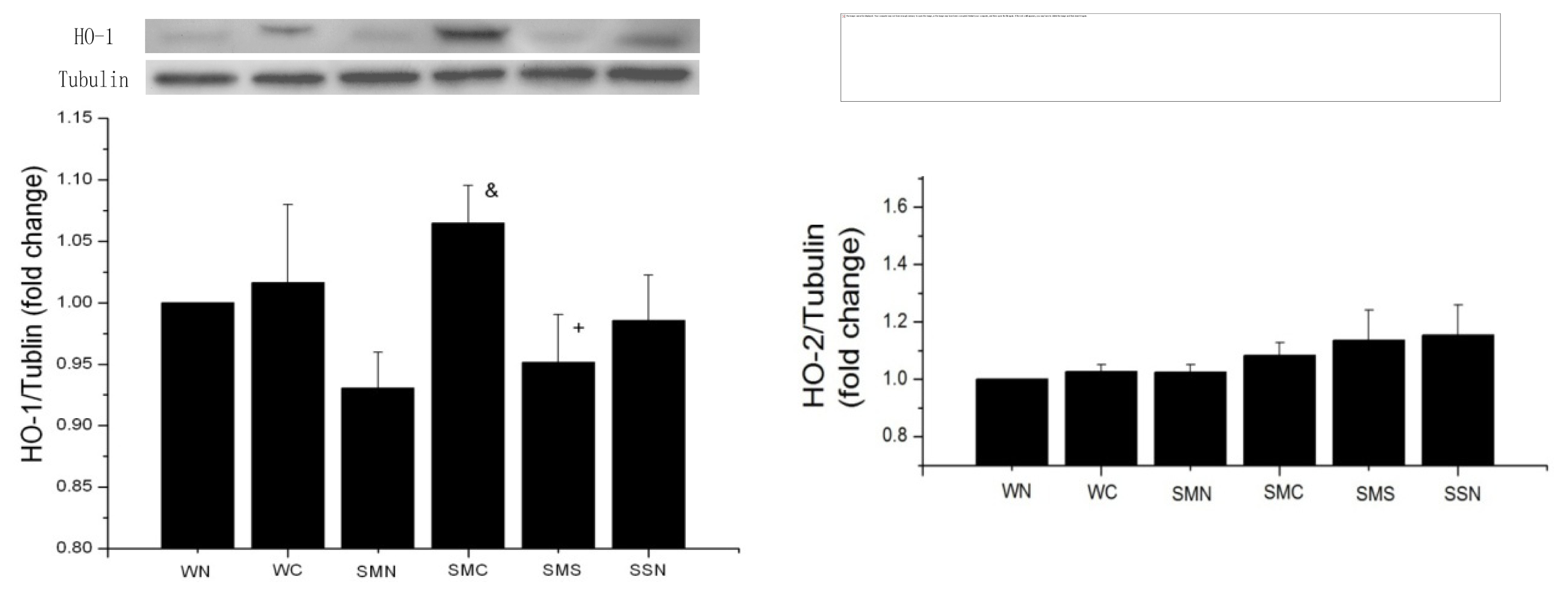
2.1.2. CoPP Upregulated HO-1 Expression in ISHR Model
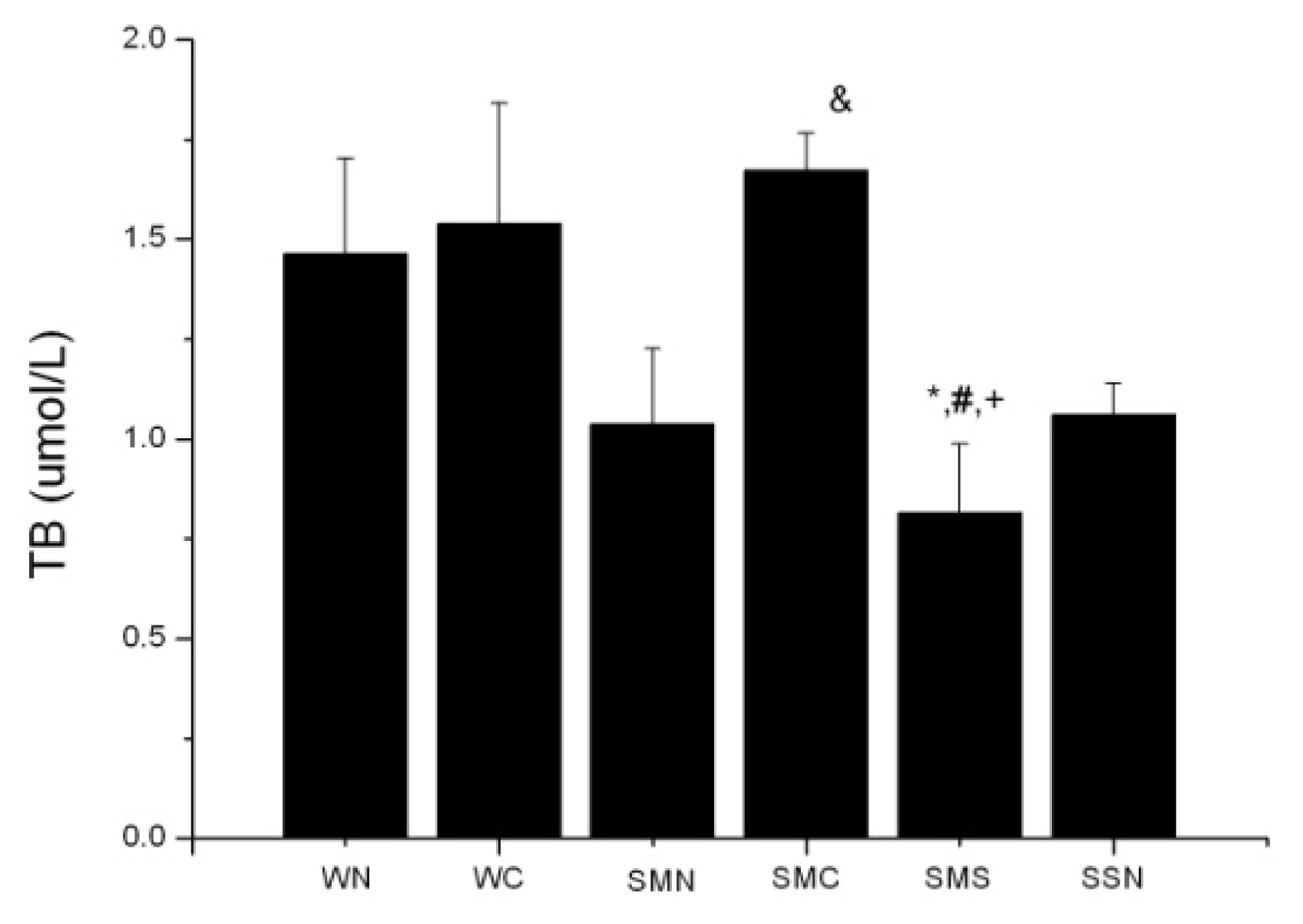
2.1.3. CoPP Treatment Enhanced Serum Total Bilirubin Levels
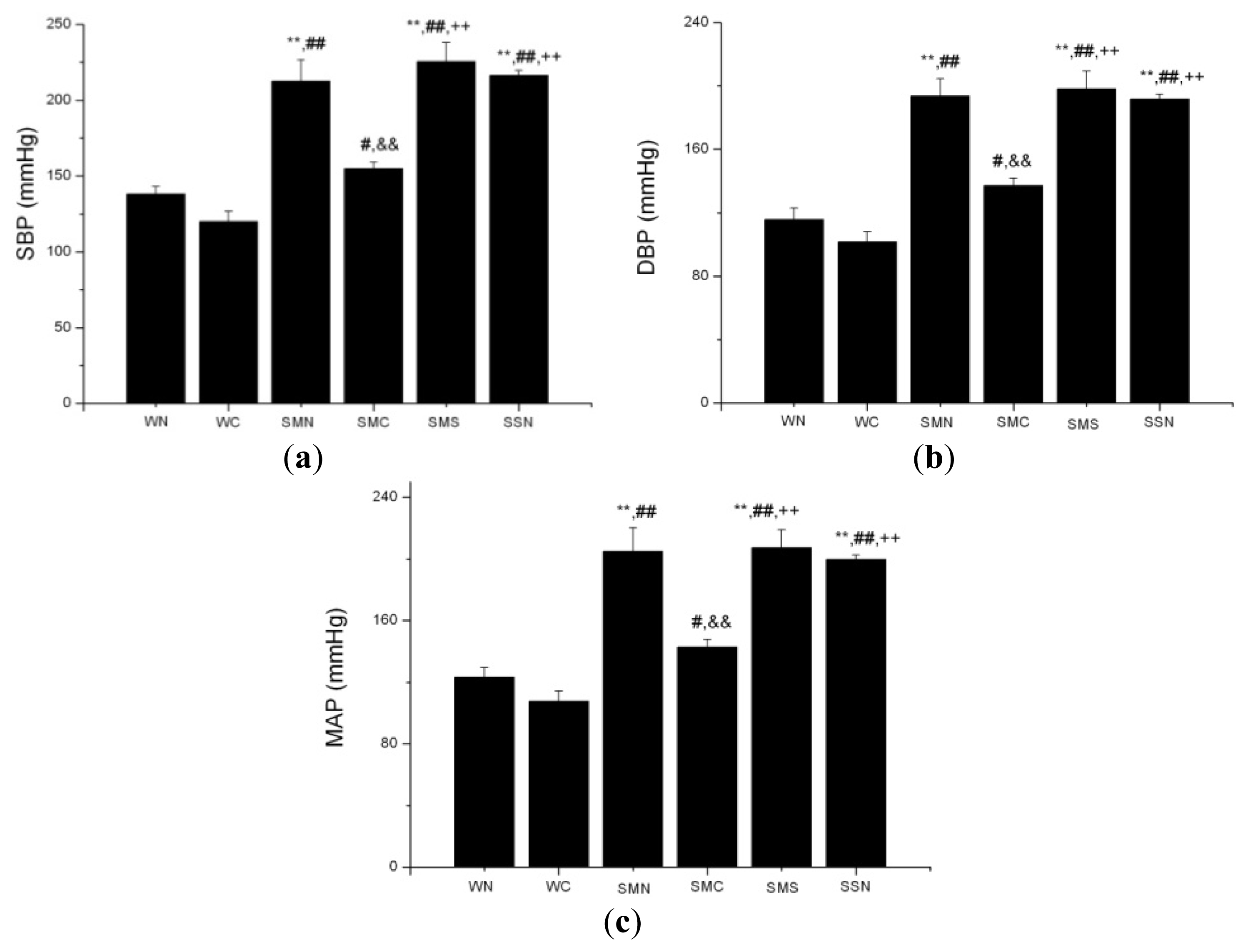
2.1.4. CoPP Treatment Reduced Blood Pressure in SHR
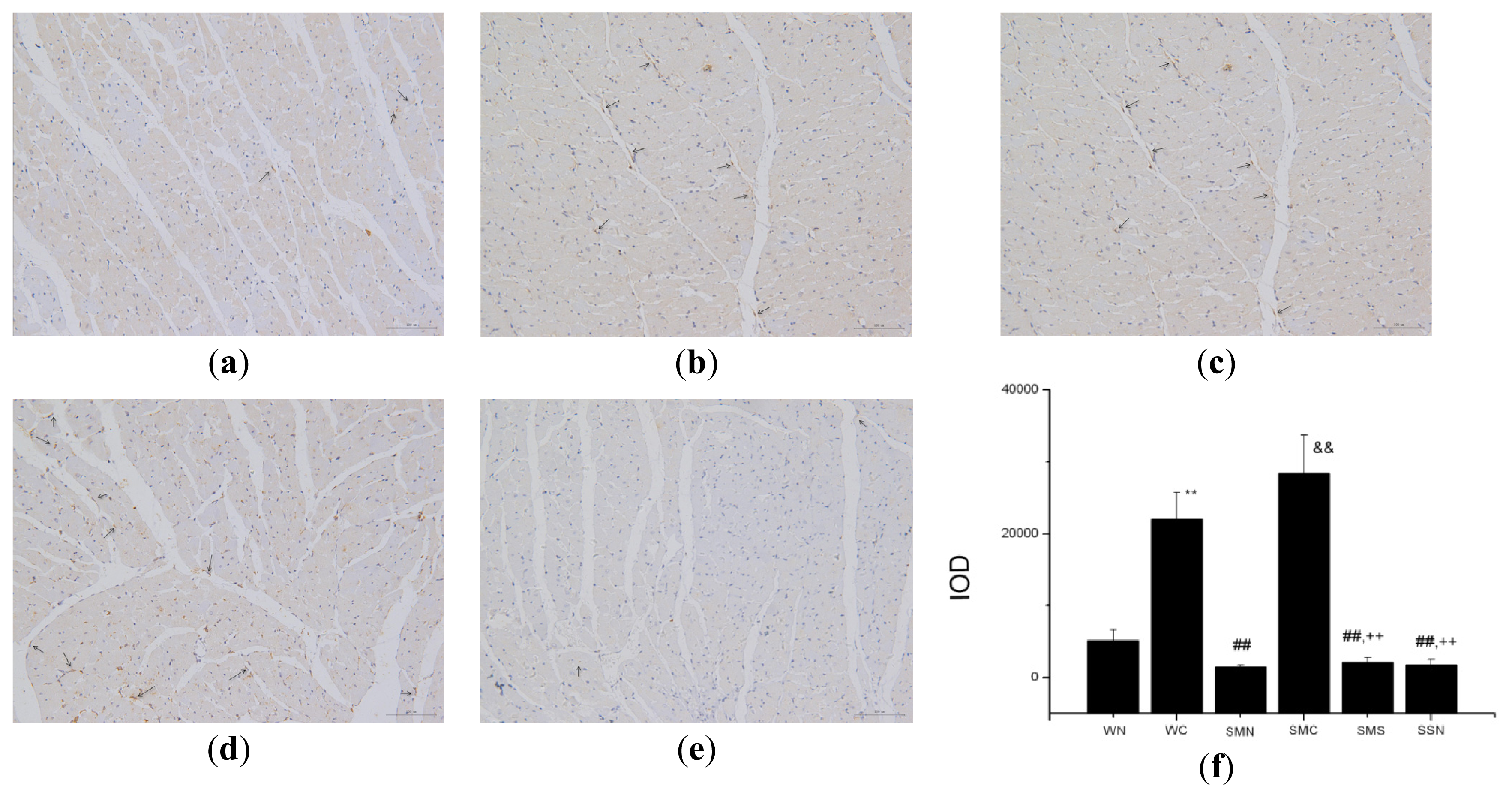
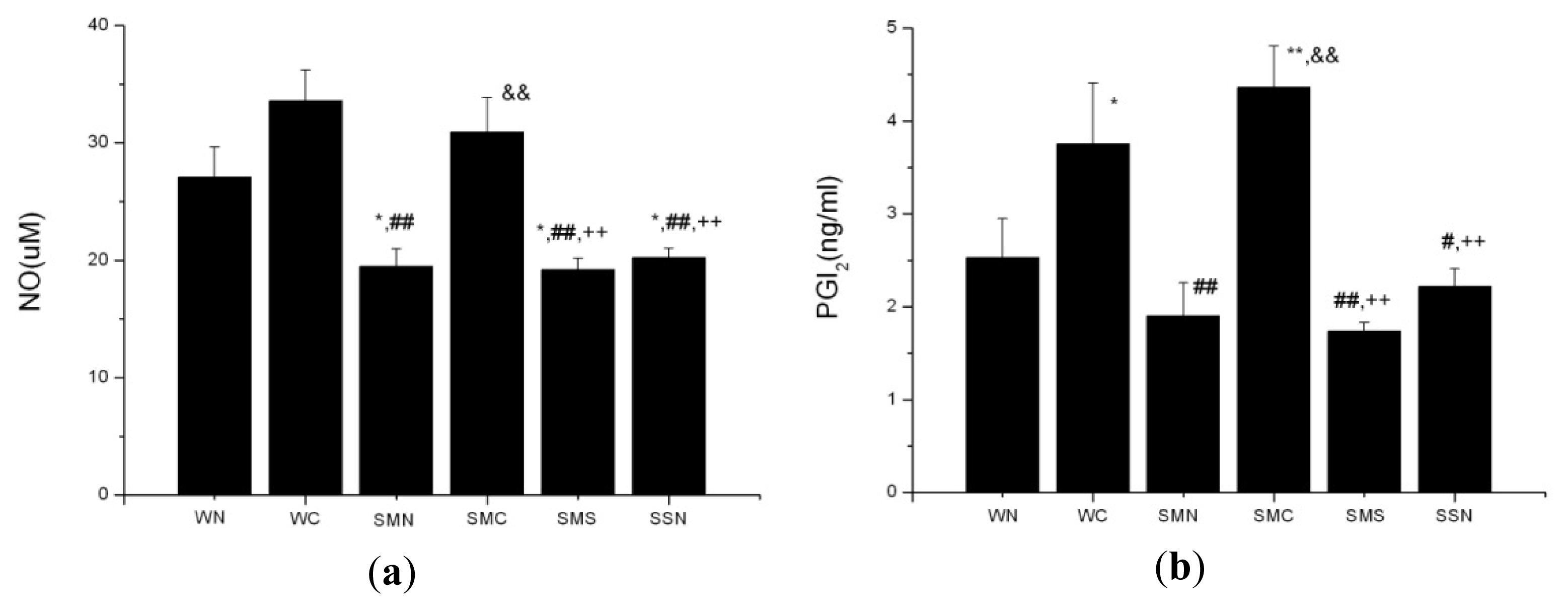
2.1.5. CoPP Treatment Ameliorated Endothelial Dysfunction
2.1.6. CoPP Treatment Diminished Infarct and Peri-Infarct Area in ISHR Model
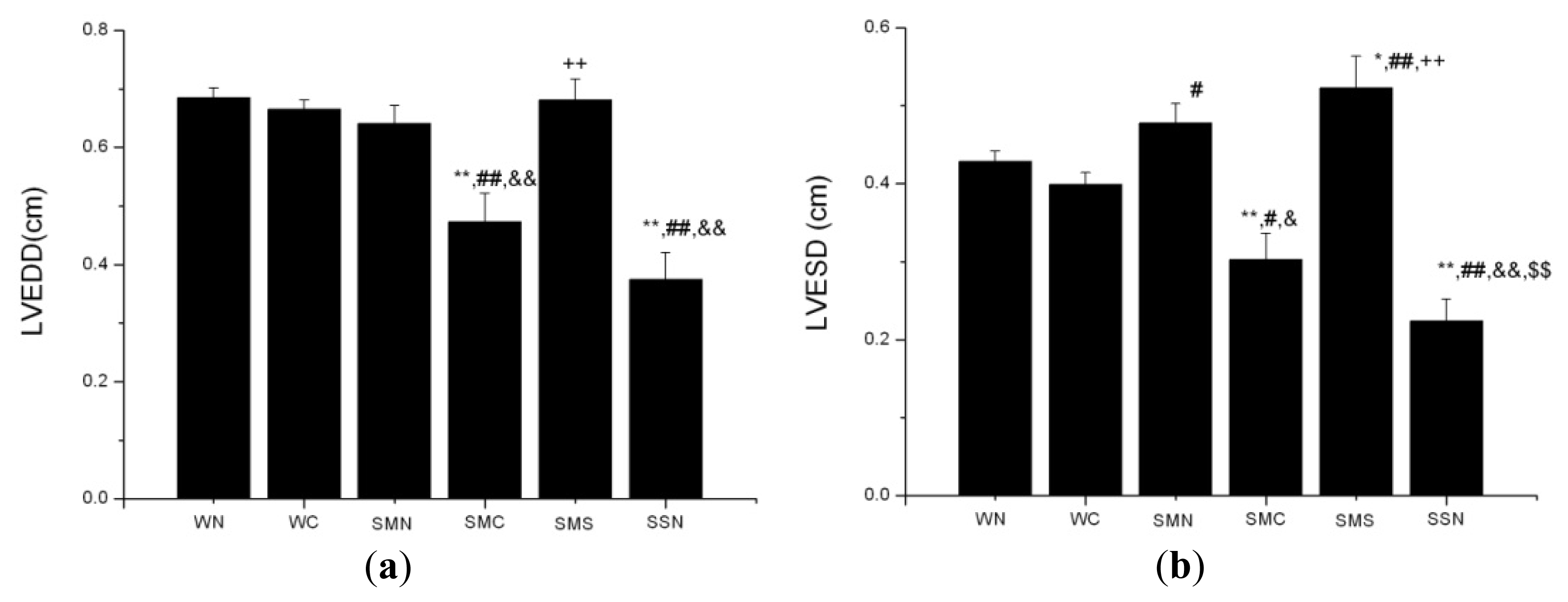
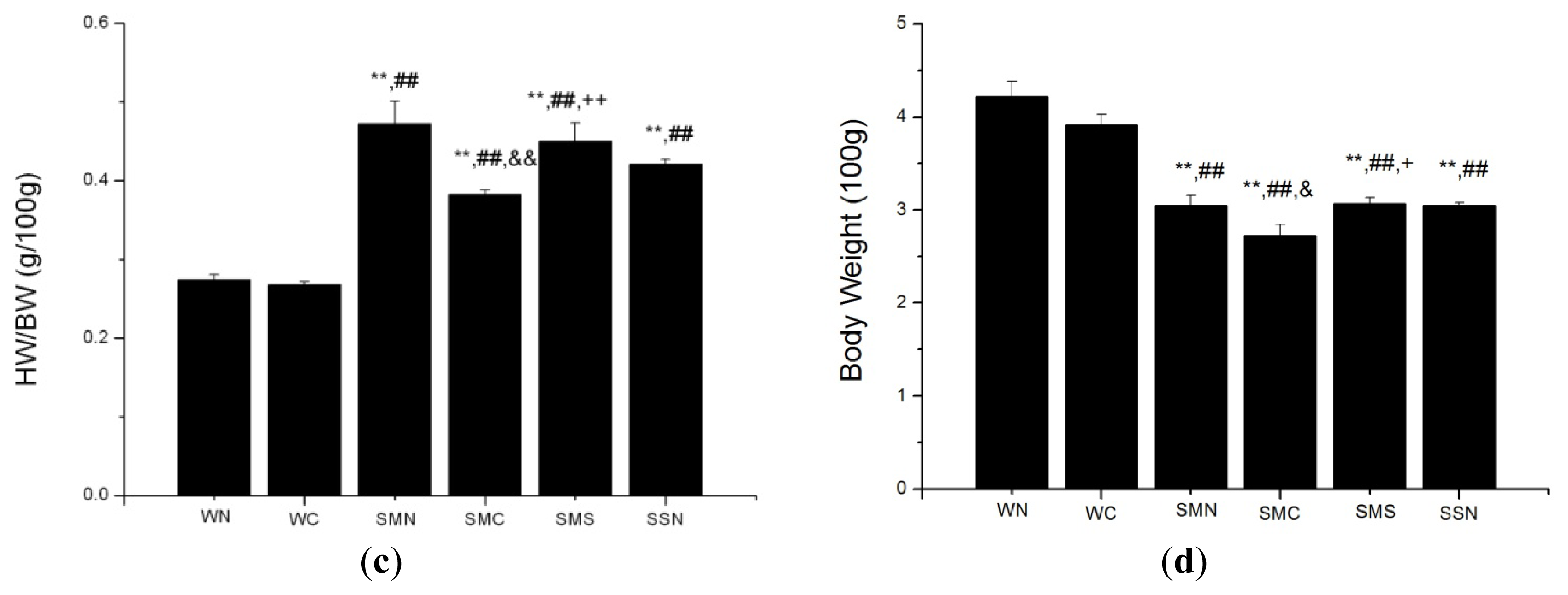
2.1.7. CoPP Treatment Inhibited Ventricular Remodeling of Post-Infarction in SHR
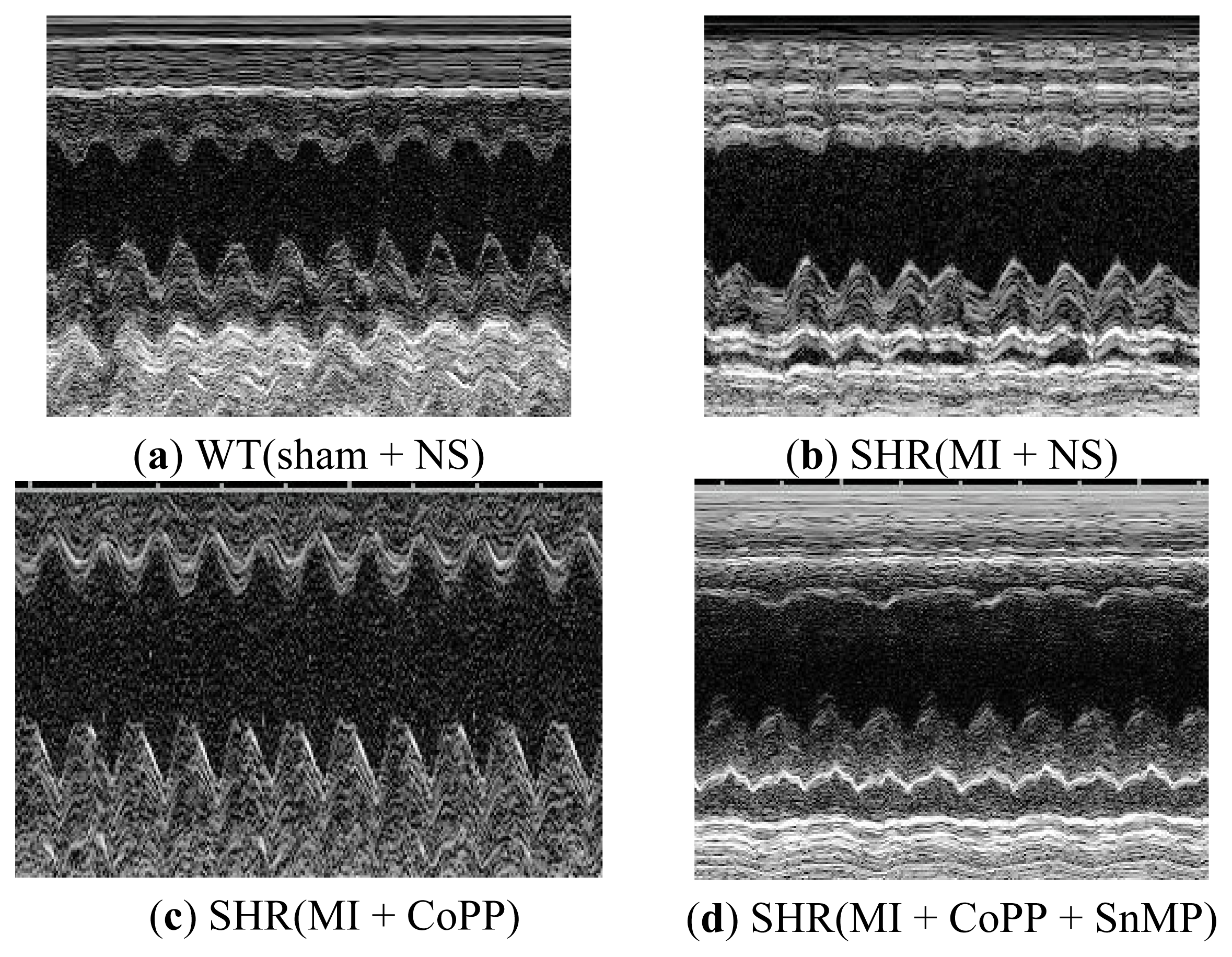
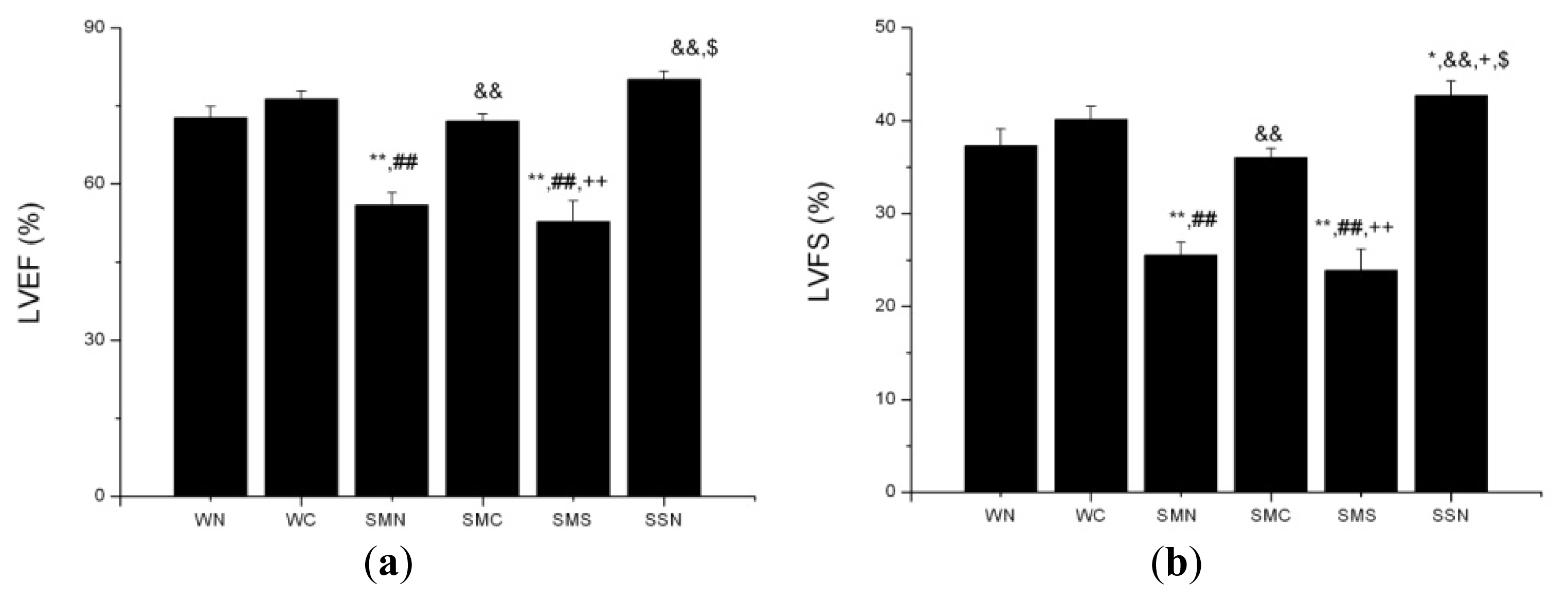
2.1.8. CoPP Treatment Improved Cardiac Function of Post-Infarction in SHR
2.1.9. CoPP Treatment Suppressed Inflammatory Cytokines Levels
2.2. Discussion
2.2.1. HO-1 Attenuation of Blood Pressure and Amelioration of Endothelial Dysfunction
2.2.2. HO-1 Modulation of Post-Infarction Pathological LV Remodeling
2.2.3. HO-1 Amelioration of Hypertensive Post-Infarction Cardiac Function
2.2.4. HO-1 Inhibition of Inflammatory Reaction
3. Experimental Section
3.1. Animal Treatment
3.2. Blood Pressure Measurements
3.3. Echocardiograph
3.4. Cardiac Catheterization
3.5. Morphology and Immunohistochemistry
3.6. Serum TB, Glu, CRP, PGI2, IL-6, NO
3.7. Western Blot
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Abbreviations
| HO-1 | heme oxygenase 1 |
| CoPP | Co(III) Protoporphyrin IX Chloride |
| SnMP | Tin Mesoporphyrin IX Dichloride |
| TB | total bilirubin |
| WT | wistar rat |
| SHR | spontaneous hypertensive rat |
| SBP | systolic blood pressure |
| DBP | diastolic blood pressure |
| MAP | mean arterial pressure |
| LVEDD | left ventricular end-diastolic diameter |
| LVESD | left ventricular end-systolic diameter |
| LVSP | left ventricular systolic pressure |
| LVEF | left ventricular ejection fraction |
| LVFS | left ventricular fraction shortening |
| HW/BW | heart weight/body weight |
| TB | total bilirubin |
| PGI2 | prostacyclin |
| NO | nitric oxide |
| MI | myocardial infarction |
| NS | normal saline |
| UCG | echocardiography |
Conflict of Interest
References
- Rembek, M.; Goch, A.; Goch, J. The clinical course of acute st-elevation myocardial infarction in patients with hypertension. Kardiol. Pol 2010, 68, 157–163. [Google Scholar]
- Peterson, S.J.; Frishman, W.H. Targeting heme oxygenase: Therapeutic implications for diseases of the cardiovascular system. Cardiol. Rev 2009, 17, 99–111. [Google Scholar]
- Wang, C.Y.; Chau, L.Y. Heme oxygenase-1 in cardiovascular diseases: Molecular mechanisms and clinical perspectives. Chang Gung Med. J 2010, 33, 13–24. [Google Scholar]
- Ferenbach, D.A.; Kluth, D.C.; Hughes, J. Hemeoxygenase-1 and renal ischaemia-reperfusion injury. Nephron. Exp. Nephrol 2010, 115, e33–e37. [Google Scholar]
- Li, J.; Zhou, Z.; Jiang, D.J.; Li, D.; Tan, B.; Liu, H.; Li, Y.J. Reduction of no- and edhf-mediated vasodilatation in hypertension: Role of asymmetric dimethylarginine. Clin. Exp. Hypertens 2007, 29, 489–501. [Google Scholar]
- Boglioni, F.V.; Metra, M.; Locati, M.; Nodari, S.; Bontempi, L.; Garbellini, M.; Doni, A.; Peri, G.; Mantovani, A. Role of inflammation mediators in the pathogenesis of heart failure. Ital. Heart J. Suppl 2001, 2, 628–633. [Google Scholar]
- Choi, H.C.; Lee, K.Y.; Lee, D.H.; Kang, Y.J. Heme oxygenase-1 induced by aprotinin inhibits vascular smooth muscle cell proliferation through cell cycle arrest in hypertensive rats. Korean J. Physiol. Pharmacol 2009, 13, 309–313. [Google Scholar]
- Ndisang, J.F.; Jadhav, A. Upregulating the heme oxygenase system suppresses left ventricular hypertrophy in adult spontaneously hypertensive rats for 3 months. J. Card. Fail 2009, 15, 616–628. [Google Scholar]
- Jadhav, A.; Ndisang, J.F. Heme arginate suppresses cardiac lesions and hypertrophy in deoxycorticosterone acetate-salt hypertension. Exp. Biol. Med. (Maywood) 2009, 234, 764–778. [Google Scholar]
- Jadhav, A.; Torlakovic, E.; Ndisang, J.F. Interaction among heme oxygenase, nuclear factor-kappab, and transcription activating factors in cardiac hypertrophy in hypertension. Hypertension 2008, 52, 910–917. [Google Scholar]
- Wang, G.; Hamid, T.; Keith, R.J.; Zhou, G.; Partridge, C.R.; Xiang, X.; Kingery, J.R.; Lewis, R.K.; Li, Q.; Rokosh, D.G.; et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 2010, 121, 1912–1925. [Google Scholar]
- Zeng, B.; Lin, G.; Ren, X.; Zhang, Y.; Chen, H. Overexpression of ho-1 on mesenchymal stem cells promotes angiogenesis and improves myocardial function in infarcted myocardium. J. Biomed. Sci 2010, 17, 80. [Google Scholar]
- Kanellakis, P.; Pomilio, G.; Agrotis, A.; Gao, X.; Du, X.J.; Curtis, D.; Bobik, A. Darbepoetin-mediated cardioprotection after myocardial infarction involves multiple mechanisms independent of erythropoietin receptor-common beta-chain heteroreceptor. Br. J. Pharmacol 2010, 160, 2085–2096. [Google Scholar]
- Penumathsa, S.V.; Koneru, S.; Samuel, S.M.; Maulik, G.; Bagchi, D.; Yet, S.F.; Menon, V.P.; Maulik, N. Strategic targets to induce neovascularization by resveratrol in hypercholesterolemic rat myocardium: Role of caveolin-1, endothelial nitric oxide synthase, hemeoxygenase-1, and vascular endothelial growth factor. Free Radic. Biol. Med 2008, 45, 1027–1034. [Google Scholar]
- Koneru, S.; Penumathsa, S.V.; Thirunavukkarasu, M.; Zhan, L.; Maulik, N. Thioredoxin-1 gene delivery induces heme oxygenase-1 mediated myocardial preservation after chronic infarction in hypertensive rats. Am. J. Hypertens 2009, 22, 183–190. [Google Scholar]
- Cao, J.; Sodhi, K.; Puri, N.; Monu, S.R.; Rezzani, R.; Abraham, N.G. High fat diet enhances cardiac abnormalities in shr rats: Protective role of heme oxygenase-adiponectin axis. Diabetol. Metab. Syndr 2011, 3, 37. [Google Scholar]
- Cao, J.; Drummond, G.; Inoue, K.; Sodhi, K.; Li, X.Y.; Omura, S. Upregulation of heme oxygenase-1 combined with increased adiponectin lowers blood pressure in diabetic spontaneously hypertensive rats through a reduction in endothelial cell dysfunction, apoptosis and oxidative stress. Int. J. Mol. Sci 2008, 9, 2388–2406. [Google Scholar]
- Papadakis, J.A.; Ganotakis, E.S.; Jagroop, I.A.; Mikhailidis, D.P.; Winder, A.F. Effect of hypertension and its treatment on lipid, lipoprotein (a), fibrinogen,and bilirubin levels in patients referred for dyslipidemia. Am. J. Hypertens 1999, 12, 673–681. [Google Scholar]
- Porteri, E.; Rodella, L.F.; Rezzani, R.; Rizzoni, D.; Paiardi, S.; de Ciuceis, C.; Boari, G.E.M.; Foglio, E.; Favero, G.; Rizzardi, N. Role of heme oxygenase in modulating endothelial function in mesenteric small resistance arteries of spontaneously hypertensive rats. Clin. Exp. Hypertens 2009, 31, 560–571. [Google Scholar]
- Jeon, E.M.; Choi, H.C.; Lee, K.Y.; Chang, K.C.; Kang, Y.J. Hemin inhibits hypertensive rat vascular smooth muscle cell proliferation through regulation of cyclin d and p21. Arch. Pharm. Res 2009, 32, 375–382. [Google Scholar]
- Elmarakby, A.A.; Faulkner, J.; Posey, S.P.; Sullivan, J.C. Induction of hemeoxygenase-1 attenuates the hypertension and renal inflammation in spontaneously hypertensive rats. Pharmacol. Res 2010, 62, 400–407. [Google Scholar]
- Torok, J. Participation of nitric oxide in different models of experimental hypertension. Physiol. Res 2008, 57, 813–825. [Google Scholar]
- Cao, J.; Inoue, K.; Li, X.; Drummond, G.; Abraham, N.G. Physiological significance of heme oxygenase in hypertension. Int. J. Biochem. Cell Biol 2009, 41, 1025–1033. [Google Scholar]
- Lee, C.Y.; Yen, M.H. Nitric oxide and carbon monoxide, collaborative and competitive regulators of hypertension. Chang Gung Med. J 2009, 32, 12–21. [Google Scholar]
- Mizuno, Y.; Jacob, R.F.; Mason, R.P. Advances in pharmacologic modulation of nitric oxide in hypertension. Curr. Cardiol. Rep 2010, 12, 472–480. [Google Scholar]
- Zhang, C.; Hein, T.W.; Wang, W.; Miller, M.W.; Fossum, T.W.; McDonald, M.M.; Humphrey, J.D.; Kuo, L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension 2004, 44, 935–943. [Google Scholar]
- Yuhki, K.; Kojima, F.; Kashiwagi, H.; Kawabe, J.; Fujino, T.; Narumiya, S.; Ushikubi, F. Roles of prostanoids in the pathogenesis of cardiovascular diseases: Novel insights from knockout mouse studies. Pharmacol. Ther 2011, 129, 195–205. [Google Scholar]
- Pavlopoulos, H.; Nihoyannopoulos, P. The constellation of hypertensive heart disease. Hell. J. Cardiol 2008, 49, 92–99. [Google Scholar]
- Oliveira, S.A., Jr; Okoshi, K.; Lima-Leopoldo, A.P.; Leopoldo, A.S.; Campos, D.H.; Martinez, P.F.; Okoshi, M.P.; Padovani, C.R.; Pai-Silva, M.D.; Cicogna, A.C. Nutritional and cardiovascular profiles of normotensive and hypertensive rats kept on a high fat diet. Arq. Bras. Cardiol. 2009, 93, 526–533. [Google Scholar]
- Fuchs, M.; Drexler, H. Mechanisms of inflammation in heart failure. Herz 2004, 29, 782–787. [Google Scholar]
- Ong, K.L.; Tso, A.W.; Xu, A.; Law, L.S.; Li, M.; Wat, N.M.; Rye, K.A.; Lam, T.H.; Cheung, B.M.; Lam, K.S. Evaluation of the combined use of adiponectin and c-reactive protein levels as biomarkers for predicting the deterioration in glycaemia after a median of 5.4 years. Diabetologia 2011, 54, 2552–2560. [Google Scholar]
- Torun, D.; Ozelsancak, R.; Yigit, F.; Micozkadioglu, H. Increased inflammatory markers are associated with obesity and not with target organ damage in newly diagnosed untreated essential hypertensive patients. Clin. Exp. Hypertens 2012, 34, 171–175. [Google Scholar]
- Yang, J.; Wang, J.; Zhu, S.; Chen, X.; Wu, H.; Yang, D.; Zhang, J. C-reactive protein augments hypoxia-induced apoptosis through mitochondrion-dependent pathway in cardiac myocytes. Mol. Cell Biochem 2008, 310, 215–226. [Google Scholar]
- Takahashi, T.; Anzai, T.; Kaneko, H.; Mano, Y.; Anzai, A.; Nagai, T.; Kohno, T.; Maekawa, Y.; Yoshikawa, T.; Fukuda, K.; et al. Increased c-reactive protein expression exacerbates left ventricular dysfunction and remodeling after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol 2010, 299, H1795–H1804. [Google Scholar]
- Pepys, M.B.; Hirschfield, G.M.; Tennent, G.A.; Gallimore, J.R.; Kahan, M.C.; Bellotti, V.; Hawkins, P.N.; Myers, R.M.; Smith, M.D.; Polara, A.; et al. Targeting c-reactive protein for the treatment of cardiovascular disease. Nature 2006, 440, 1217–1221. [Google Scholar]
- Gryglewski, R.J. Prostacyclin among prostanoids. Pharmacol. Rep 2008, 60, 3–11. [Google Scholar]
- Rockman, H.A.; Ross, R.S.; Harris, A.N.; Knowlton, K.U.; Steinhelper, M.E.; Field, L.J.; Ross, J., Jr; Chien, K.R. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 1991, 88, 8277–8281. [Google Scholar]
- Tei, C.; Nishimura, R.A.; Seward, J.B.; Tajik, A.J. Noninvasive doppler-derived myocardial performance index: Correlation with simultaneous measurements of cardiac catheterization measurements. J. Am. Soc. Echocardiogr 1997, 10, 169–178. [Google Scholar]
- Weisfeldt, M.L.; Scully, H.E.; Frederiksen, J.; Rubenstein, J.J.; Pohost, G.M.; Beierholm, E.; Bello, A.G.; Daggett, W.M. Hemodynamic determinants of maximum negative dp/dt and periods of diastole. Am. J. Physiol 1974, 227, 613–621. [Google Scholar]
- Slama, M.; Ahn, J.; Peltier, M.; Maizel, J.; Chemla, D.; Varagic, J.; Susic, D.; Tribouilloy, C.; Frohlich, E.D. Validation of echocardiographic and doppler indexes of left ventricular relaxation in adult hypertensive and normotensive rats. Am. J. Physiol. Heart Circ. Physiol 2005, 289, H1131–H1136. [Google Scholar]
- Grousset, C.; Menasche, P.; Apstein, C.S.; Mouas, C.; Marotte, F.; Piwnica, A. Protective effects of cardioplegia on diastolic function of hypertrophied rat hearts after hypothermic ischaemic arrest. Eur. Heart J 1984, 5, 347–353. [Google Scholar]
- Cooper, D.J.; Herbertson, M.J.; Werner, H.A.; Walley, K.R. Bicarbonate does not increase left ventricular contractility during l-lactic acidemia in pigs. Am. Rev. Respir. Dis 1993, 148, 317–322. [Google Scholar]
- Mahler, F.; Ross, J., Jr; O’Rourke, R.A.; Covell, J.W. Effects of changes in preload, afterload and inotropic state on ejection and isovolumic phase measures of contractility in the conscious dog. Am. J. Cardiol. 1975, 35, 626–634. [Google Scholar]
- Savoia, C.; Sada, L.; Zezza, L.; Pucci, L.; Lauri, F.M.; Befani, A.; Alonzo, A.; Volpe, M. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int. J. Hypertens 2011, 2011, 281240. [Google Scholar]
- Celik, A.; Koc, F.; Kadi, H.; Ceyhan, K.; Erkorkmaz, U. Inflammation is related to unbalanced cardiac autonomic functions in hypertension: An observational study. Anadolu. Kardiyol. Derg 2012, 12, 233–240. [Google Scholar]
- Barnes, T.C.; Anderson, M.E.; Moots, R.J. The many faces of interleukin-6: The role of il-6 in inflammation, vasculopathy, and fibrosis in systemic sclerosis. Int. J. Rheumatol 2011, 2011, 721608. [Google Scholar]
- Kurdi, M.; Randon, J.; Cerutti, C.; Bricca, G. Increased expression of il-6 and lif in the hypertrophied left ventricle of tgr(mren2)27 and shr rats. Mol. Cell Biochem 2005, 269, 95–101. [Google Scholar]
- Sturgis, L.C.; Cannon, J.G.; Schreihofer, D.A.; Brands, M.W. The role of aldosterone in mediating the dependence of angiotensin hypertension on il-6. Am. J. Physiol. Regul. Integr. Comp. Physiol 2009, 297, R1742–R1748. [Google Scholar]
- Gabriel, A.S.; Martinsson, A.; Wretlind, B.; Ahnve, S. Il-6 levels in acute and post myocardial infarction: Their relation to crp levels, infarction size, left ventricular systolic function, and heart failure. Eur. J. Intern. Med 2004, 15, 523–528. [Google Scholar]
- Chamarthi, B.; Williams, G.H.; Ricchiuti, V.; Srikumar, N.; Hopkins, P.N.; Luther, J.M.; Jeunemaitre, X.; Thomas, A. Inflammation and hypertension: The interplay of interleukin-6, dietary sodium, and the renin-angiotensin system in humans. Am. J. Hypertens 2011, 24, 1143–1148. [Google Scholar]
- Cheung, B.M.; Ong, K.L.; Tso, A.W.; Leung, R.Y.; Xu, A.; Cherny, S.S.; Sham, P.C.; Lam, T.H.; Lam, K.S. C-reactive protein as a predictor of hypertension in the hong kong cardiovascular risk factor prevalence study (crisps) cohort. J. Hum. Hypertens 2012, 26, 108–116. [Google Scholar]
- Li, X.; Yang, G.; Zhao, G.; Wu, B.; Edin, M.L.; Zeldin, D.C.; Wang, D.W. Rosuvastatin attenuates the elevation in blood pressure induced by overexpression of human c-reactive protein. Hypertens. Res 2011, 34, 869–875. [Google Scholar]
- Khan, H.A.; Alhomida, A.S.; Sobki, S.H.; Moghairi, A.A.; Koronki, H.E. Blood cell counts and their correlation with creatine kinase and c-reactive protein in patients with acute myocardial infarction. Int. J. Clin. Exp. Med 2012, 5, 50–55. [Google Scholar]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.X.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y.B.; et al. Oxidant stress form nitric oxide synthase-3 uncoupling stimulateds cardiac pathologic remodeling from chronic pressure load. J. Clin. Invest 2005, 115, 1221–1231. [Google Scholar]
- Abraham, N.G.; Kushida, T.; McClung, J.; Weiss, M.; Quan, S.; Lafaro, R.; Darzynkiewicz, Z.; Wolin, M. Heme oxygenase-1 attenuates glucose-mediated cell growth arrest and apoptosis in human microvessel endothelial cells. Circ. Res 2003, 93, 507–514. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WTs (n = 20) | SHRs (n = 40) | p | |
|---|---|---|---|
| HR | 383.70 ± 14.174 | 379.69 ± 37.316 | 0.741 |
| SBP | 143.50 ± 198.60 | 198.60 ± 18.210 | <0.001 |
| MAP | 124.60 ± 6.786 | 170.90 ± 19.503 | <0.001 |
| DBP | 115.20 ± 6.374 | 157.24 ± 21.121 | <0.001 |
| WTs (n = 20) | SHRs (n = 40) | p | |
|---|---|---|---|
| LVEDD (cm) | 0.5888 ± 0.0469 | 0.4060 ± 0.0457 | 0.004 |
| LVESD (cm) | 0.3563 ± 0.03758 | 0.2240 ± 0.04486 | 0.008 |
| LVFS (%) | 40.150 ± 5.1094 | 44.640 ± 3.9339 | 0.189 |
| LVEF (%) | 76.200 ± 0.0535 | 81.560 ± 0.0744 | 0.144 |
| Groups | Masson (macroscopic) | Masson (×200) | Infarct area (HE ×200) | Peri-infarct area (HE ×200) |
|---|---|---|---|---|
| WN |  |  |  | |
| SMN |  |  |  |  |
| SMC |  |  |  |  |
| SMS |  |  |  |  |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, T.-m.; Li, J.; Liu, L.; Fan, L.; Li, X.-y.; Wang, Y.-t.; Abraham, N.G.; Cao, J. Effects of Heme Oxygenase-1 Upregulation on Blood Pressure and Cardiac Function in an Animal Model of Hypertensive Myocardial Infarction. Int. J. Mol. Sci. 2013, 14, 2684-2706. https://doi.org/10.3390/ijms14022684
Chen T-m, Li J, Liu L, Fan L, Li X-y, Wang Y-t, Abraham NG, Cao J. Effects of Heme Oxygenase-1 Upregulation on Blood Pressure and Cardiac Function in an Animal Model of Hypertensive Myocardial Infarction. International Journal of Molecular Sciences. 2013; 14(2):2684-2706. https://doi.org/10.3390/ijms14022684
Chicago/Turabian StyleChen, Tian-meng, Jian Li, Lin Liu, Li Fan, Xiao-ying Li, Yu-tang Wang, Nader G. Abraham, and Jian Cao. 2013. "Effects of Heme Oxygenase-1 Upregulation on Blood Pressure and Cardiac Function in an Animal Model of Hypertensive Myocardial Infarction" International Journal of Molecular Sciences 14, no. 2: 2684-2706. https://doi.org/10.3390/ijms14022684
APA StyleChen, T.-m., Li, J., Liu, L., Fan, L., Li, X.-y., Wang, Y.-t., Abraham, N. G., & Cao, J. (2013). Effects of Heme Oxygenase-1 Upregulation on Blood Pressure and Cardiac Function in an Animal Model of Hypertensive Myocardial Infarction. International Journal of Molecular Sciences, 14(2), 2684-2706. https://doi.org/10.3390/ijms14022684