Effects of Aging and Hypercholesterolemia on Oxidative Stress and DNA Damage in Bone Marrow Mononuclear Cells in Apolipoprotein E-deficient Mice


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
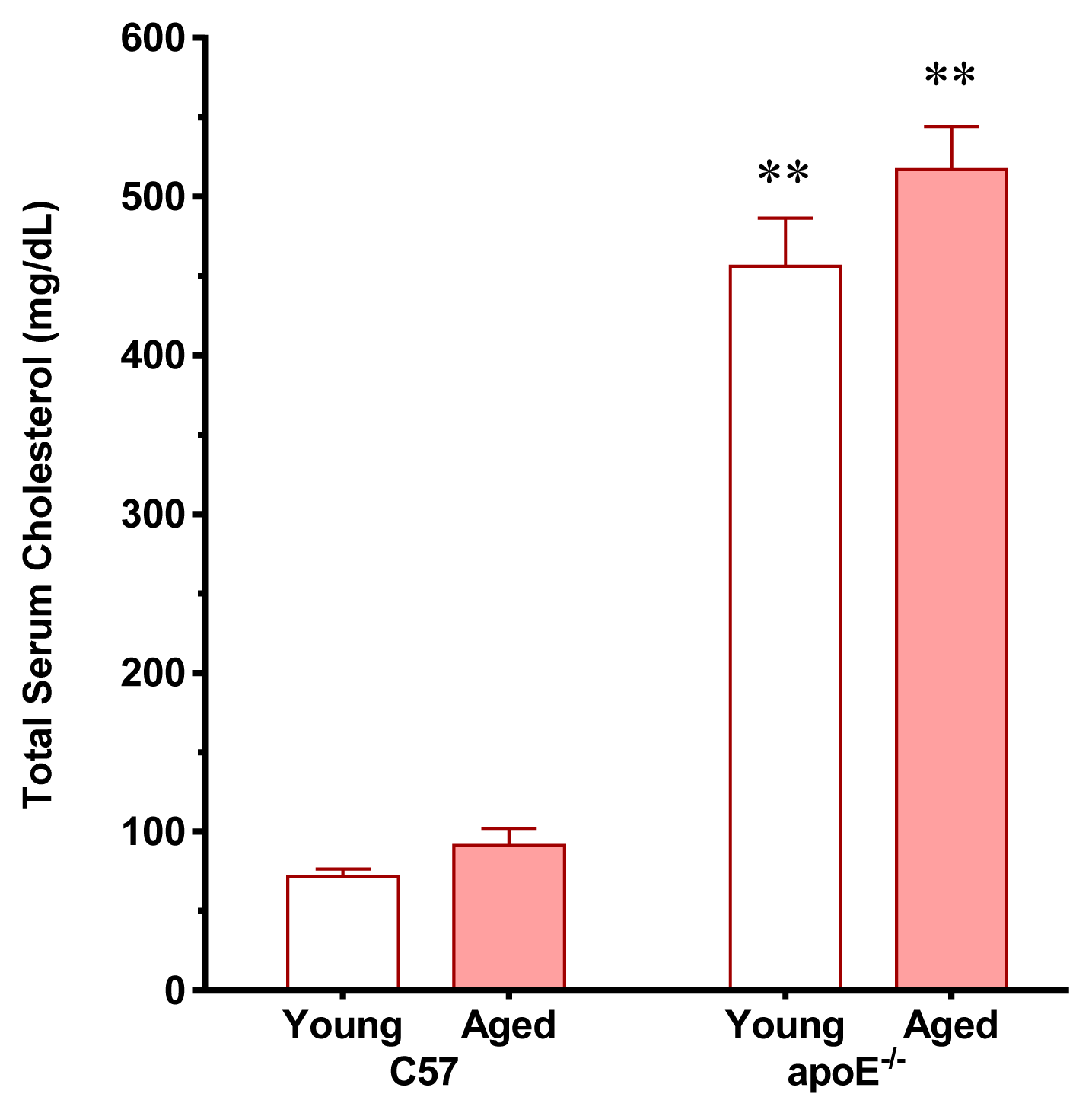
2.1. Vascular Senescence and Lipid Deposition
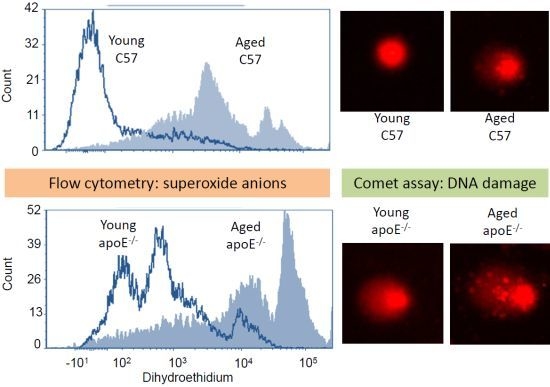
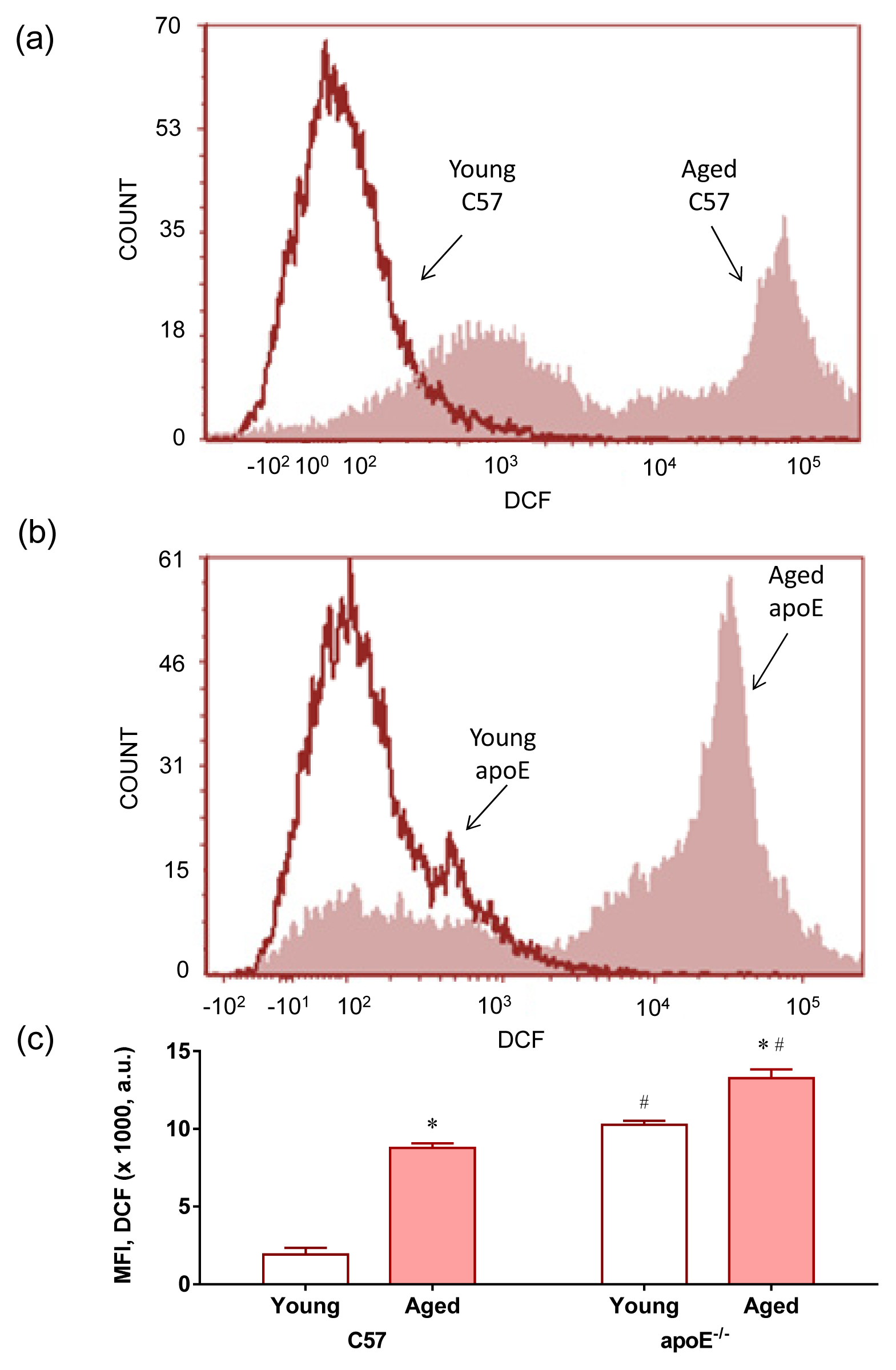
2.2. Reactive Oxygen Species (ROS)
2.3. Analysis of DNA Damage (Comet Assay)
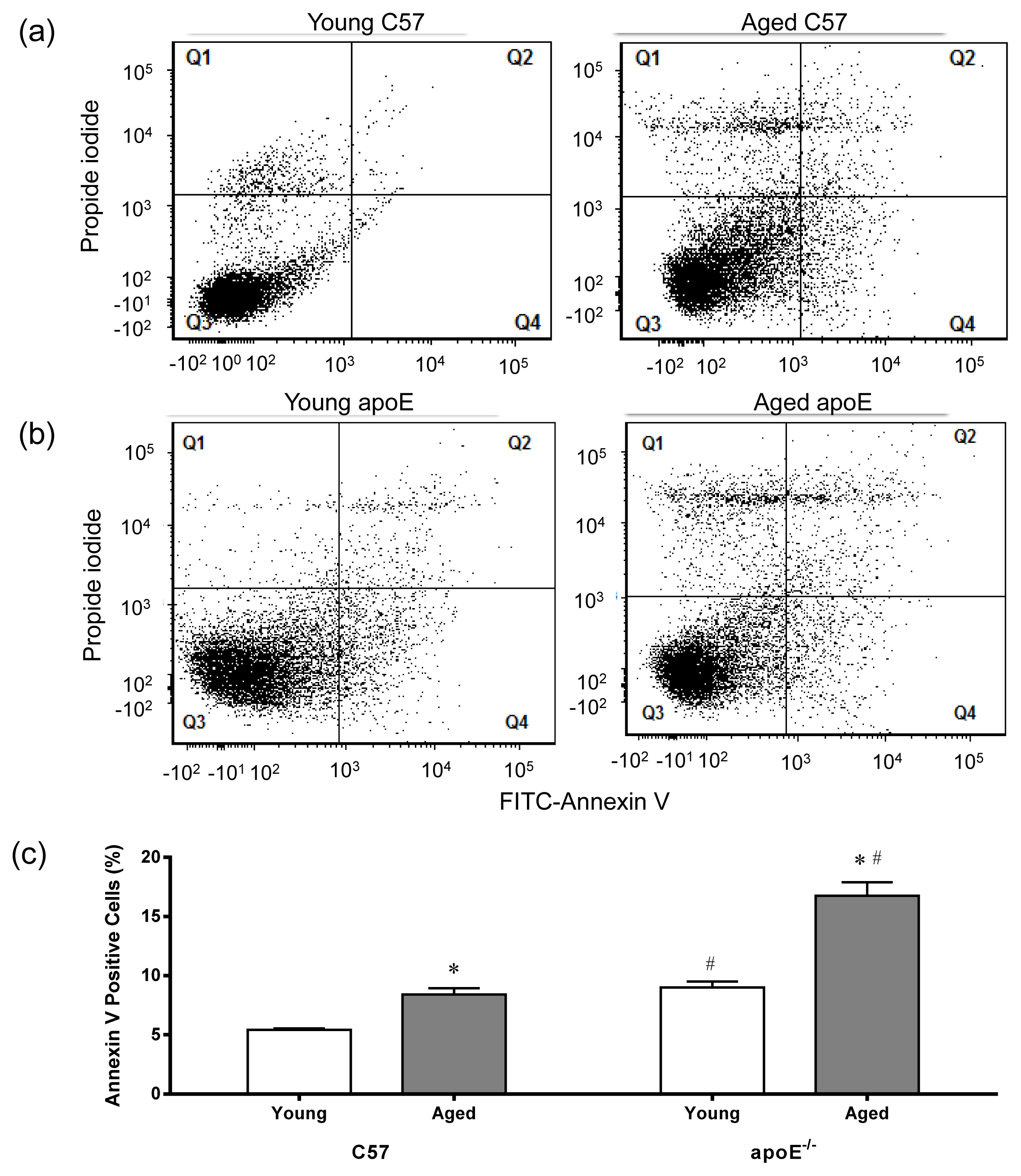
2.4. Apoptosis
3. Experimental Section
3.1. Animals
3.2. Analysis of Plasma Cholesterol and Glucose
3.3. Analysis of Senescence and Vascular Lipid Deposition
3.4. Isolation of Bone Marrow Mononuclear Bone Marrow Cells for Comet Assay and Flow Cytometry
3.5. Measurement of DNA Damage by the Comet Assay
3.6. Measurement of Cytoplasmatic Reactive Oxygen Species by DHE and DCFs
3.7. Apoptosis in MNC
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med 1999, 340, 115–126. [Google Scholar]
- Vasquez, E.C.; Peotta, V.A.; Gava, A.L.; Pereira, T.M.; Meyrelles, S.S. Cardiac and vascular phenotypes in the apolipoprotein E-deficient mouse. J. Biomed. Sci. 2012, 19. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2003, 21, 361–370. [Google Scholar]
- Bauer, V.; Sotníková, R.; Drábiková, K. Effects of reactive oxygen species and neutrophils on endothelium-dependent relaxation of rat thoracic aorta. Interdiscip. Toxicol 2011, 4, 191–197. [Google Scholar]
- McEwen, J.E.; Zimniak, P.; Mehta, J.L.; Shmookler Reis, R.J. Molecular pathology of aging and its implications for senescent coronary atherosclerosis. Curr. Opin. Cardiol 2005, 20, 399–406. [Google Scholar]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res 2007, 100, 460–473. [Google Scholar]
- Zhang, X.; Qi, R.; Xian, X.; Yang, F.; Blackstein, M.; Deng, X.; Fan, J.; Ross, C.; Karasinska, J.; Hayden, M.R.; et al. Spontaneous atherosclerosis in aged lipoprotein lipase-deficient mice with severe hypertriglyceridemia on a normal chow diet. Circ. Res 2008, 102, 250–256. [Google Scholar]
- Collins, A.R.; Lyon, C.J.; Xia, X.; Liu, J.Z.; Tangirala, R.K.; Yin, F.; Boyadjian, R.; Bikineyeva, A.; Praticò, D.; Harrison, D.G.; et al. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ. Res 2009, 104, e42–e54. [Google Scholar]
- Hasty, P.; Campisi, J.; Hoeijmakers, J.; van Steeg, H.; Vijg, J. Aging and genome maintenance: lessons from the mouse? Science 2003, 299, 1355–1359. [Google Scholar]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol 2007, 292, R18–R36. [Google Scholar]
- Vasquez, E.C.; Peotta, V.A.; Meyrelles, S.S. Cardiovascular autonomic imbalance and baroreflex dysfunction in the apolipoprotein E-deficient mouse. Cell Physiol. Biochem 2012, 29, 635–646. [Google Scholar]
- Pereira, T.M.; Nogueira, B.V.; Lima, L.C.; Porto, M.L.; Arruda, J.A.; Vasquez, E.C.; Meyrelles, S.S. Cardiac and vascular changes in elderly atherosclerotic mice: The influence of gender. Lipids Health Dis. 2010, 9, 87. [Google Scholar]
- Porto, M.L.; Lima, L.C.; Pereira, T.M.; Nogueira, B.V.; Tonini, C.L.; Campagnaro, B.P.; Meyrelles, S.S.; Vasquez, E.C. Mononuclear cell therapy attenuates atherosclerosis in apoE KO mice. Lipids Health. Dis 2011, 10, 155. [Google Scholar]
- Lima, L.C.; Porto, M.L.; Campagnaro, B.P.; Tonini, C.L.; Nogueira, B.V.; Pereira, T.M.; Vasquez, E.C.; Meyrelles, S.S. Mononuclear cell therapy reverts cuff-induced thrombosis in apolipoprotein E-deficient mice. Lipids Health. Dis 2012, 11, 96. [Google Scholar]
- Meyrelles, S.S.; Peotta, V.A.; Pereira, T.M.; Vasquez, E.C. Endothelial dysfunction in the apolipoprotein E-deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis 2011, 10, 211. [Google Scholar]
- Folkmann, J.K.; Loft, S.; Møller, P. Oxidatively damaged DNA in aging dyslipidemic apoE−/− and wild-type mice. Mutagenesis 2007, 22, 105–110. [Google Scholar]
- Dalboni, S.P.; Campagnaro, B.P.; Tonini, C.L.; Vasquez, E.C.; Meyrelles, S.S. The concurrence of hypercholesterolemia and aging promotes DNA damage in apolipoprotein E-deficient mice. Open J. Blood Dis 2012, 2, 51–55. [Google Scholar]
- Cuende, N.; Rico, L.; Herrera, C. Bone marrow mononuclear cells for the treatment of ischemic syndromes: medicinal product or cell transplantation? Stem Cells Trans. Med. 2012, 1, 403–408. [Google Scholar]
- Campagnaro, B.P.; Gava, A.L.; Meyrelles, S.S.; Vasquez, E.C. Cardiac-autonomic imbalance and baroreflex dysfunction in the renovascular angiotensin-dependent hypertensive mouse. Int. J. Hypertens. 2012, 2012. [Google Scholar] [CrossRef]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. Vasc. Biol 1994, 14, 133–140. [Google Scholar]
- Reddick, R.L.; Zhang, S.H.; Maeda, N. Atherosclerosis in mice lacking apo E: evaluation of lesional development and progression. Arterioscler. Thromb. Vasc. Biol 1994, 14, 141–147. [Google Scholar]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar]
- Jawień, J.; Nastałek, P.; Korbut, R. Mouse models of experimental atherosclerosis. J. Physiol. Pharmacol 2004, 55, 503–517. [Google Scholar]
- Goldschmidt-Clermont, P.J. Loss of bone marrow-derived vascular progenitor cells leads to inflammation and atherosclerosis. Am. Heart J 2003, 146, S5–S12. [Google Scholar]
- Kojda, G.; Harrison, D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc. Res 1999, 43, 562–571. [Google Scholar]
- Groemping, Y.; Rittinger, K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem. J 2005, 386, 401–416. [Google Scholar]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med 2005, 38, 1278–1295. [Google Scholar]
- Dikalov, S. Crosstalk between mitochondria and NADPH oxidases. Free Radic. Biol. Med 2011, 51, 1289–1301. [Google Scholar]
- Schröder, K.; Vecchione, C.; Jung, O.; Schreiber, J.G.; Shiri-Sverdlov, R.; van Gorp, P.J.; Busse, R.; Brandes, R.P. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free Radic. Biol. Med 2006, 41, 1353–1360. [Google Scholar]
- Takaya, T.; Hirata, K.; Yamashita, T.; Shinohara, M.; Sasaki, N.; Inoue, N.; Yada, T.; Goto, M.; Fukatsu, A.; Hayashi, T.; et al. A specific role for eNOS-derived reactive oxygen species in atherosclerosis progression. Arterioscler. Thromb. Vasc. Biol 2007, 27, 1632–1637. [Google Scholar]
- Jawien, J.; Gajda, M.; Wołkow, P.; Zurańska, J.; Olszanecki, R.; Korbut, R. The effect of montelukast on atherogenesis in apoE/LDLR-double knockout mice. J. Physiol. Pharmacol 2008, 59, 633–639. [Google Scholar]
- Sheehan, A.L.; Carrell, S.; Johnson, B.; Stanic, B.; Banfi, B.; Miller, F.J., Jr. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 2011, 216, 321–326. [Google Scholar]
- Zou, H.; Stoppani, E.; Volonte, D.; Galbiati, F. Caveolin-1, cellular senescence and age-related diseases. Mech. Ageing. Dev 2001, 132, 533–542. [Google Scholar]
- Law, A.; Gauthier, S.; Quirion, R. Alteration of nitric oxide synthase activity in young and aged apolipoprotein E-deficient mice. Neurobiol. Aging 2003, 24, 187–190. [Google Scholar]
- Jiang, F.; Jones, G.T.; Dusting, G.J. Failure of antioxidants to protect against angiotensin II-induced aortic rupture in aged apolipoprotein (E)-deficient mice. Br. J. Pharmacol 2007, 152, 880–890. [Google Scholar]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ. Res 2006, 99, 156–164. [Google Scholar]
- Tatarková, Z.; Kuka, S.; Račay, P.; Lehotský, J.; Dobrota, D.; Mištuna, D.; Kaplán, P. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol. Res 2011, 60, 281–289. [Google Scholar]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res 2012, 111, 245–259. [Google Scholar]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res 2004, 567, 1–61. [Google Scholar]
- Ellahueñe, M.F.; Pérez-Alzola, L.P.; Farfán-Urzua, M.; González-Hormazabal, P.; Garay, M.; Olmedo, M.I.; Last, J.A. Preliminary evaluation of DNA damage related with the smoking habit measured by the comet assay in whole blood cells. Cancer Epidemiol. Biomark. Prev 2004, 13, 1223–1229. [Google Scholar]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res 1988, 175, 184–191. [Google Scholar]
- Lee, R.F.; Steinert, S. Use of the single cell gel electrophoresis/comet assay for detecting DNA damage in aquatic (marine and freshwater) animals. Mutat. Res 2003, 544, 43–64. [Google Scholar]
- Yao, Y.; Tian, X.; Liu, X.; Shao, J.; Lv, Y. The p53-mediated apoptosis in hypercholesterolemia-induced renal injury of rats. J. Huazhong. Univ. Sci. Technol. Med. Sci 2005, 25, 408–411. [Google Scholar]
- Sevin, G.; Yasa, M.; Akcay, D.Y.; Kirkali, G.; Kerry, Z. Different responses of fluvastatin to cholesterol-induced oxidative modifications in rabbits: evidence for preventive effect against DNA damage. Cell Biochem. Funct 2012, 38, 1278–1295. [Google Scholar]
- Martinet, W.; Knaapen, M.W.; De Meyer, G.R.; Herman, A.G.; Kockx, M.M. Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ. Res 2001, 88, 733–739. [Google Scholar]
- Ronsein, G.E.; de Oliveira, M.C.; Medeiros, M.H.; Miyamoto, S.; Di Mascio, P. DNA strand breaks and base modifications induced by cholesterol hydroperoxides. Free. Radic. Res 2011, 45, 266–275. [Google Scholar]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar]
- Mercer, J.; Mahmoudi, M.; Bennett, M. DNA damage, p53, apoptosis and vascular disease. Mutat. Res 2007, 621, 75–86. [Google Scholar]
- Mercer, J.R.; Cheng, K.K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.P.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res 2010, 107, 1021–1031. [Google Scholar]
- Yu, E.; Mercer, J.; Bennett, M. Mitochondria in vascular disease. Cardiovasc. Res 2012, 95, 173–182. [Google Scholar]
- Shaposhnikov, S.; Larsson, C.; Henriksson, S.; Collins, A.; Nilsson, N. Detection of Alu sequences and mtDNA in comets using padlock probes. Mutagenesis 2006, 21, 243–247. [Google Scholar]
- Harman, D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol 1956, 11, 298–300. [Google Scholar]
- Clutton, S. The importance of oxidative stress in apoptosis. Br. Med. Bull 1997, 53, 662–668. [Google Scholar]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem 1997, 272, 20313–20316. [Google Scholar]
- Slater, T.F. Free radicals and tissue injury: fact and fiction. Br. J. Cancer. Suppl 1987, 8, 5–10. [Google Scholar]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar]
- Spiteller, G. The relation of lipid peroxidation processes with atherogenesis: A new theory on atherogenesis. Mol. Nutr. Food Res 2005, 49, 999–1013. [Google Scholar]
- Bayir, H.; Kagan, V.E. Bench-to-bedside review: Mitochondrial injury, oxidative stress and apoptosis--there is nothing more practical than a good theory. Crit. Care 2008, 12, 206. [Google Scholar]
- Papaharalambus, C.A.; Griendling, K.K. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends. Cardiovasc. Med 2007, 17, 48–54. [Google Scholar]
- Devlin, A.M.; Clark, J.S.; Reid, J.L.; Dominiczak, A.F. DNA synthesis and apoptosis in smooth muscle cells from a model of genetic hypertension. Hypertension 2000, 36, 110–115. [Google Scholar]
- Rauscher, F.M.; Goldschmidt-Clermont, P.J.; Davis, B.H.; Wang, T.; Gregg, D.; Ramaswami, P.; Pippen, A.M.; Annex, B.H.; Dong, C.; Taylor, D.A. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation 2003, 108, 457–463. [Google Scholar]
- Thorin-Trescases, N.; Voghel, G.; Gendron, M.E.; Krummen, S.; Farhat, N.; Drouin, A.; Perrault, L.P.; Thorin, E. Pathological aging of the vascular endothelium: are endothelial progenitor cells the sentinels of the cardiovascular system? Can. J. Cardiol 2005, 21, 1019–1024. [Google Scholar]
- Goldschmidt-Clermont, P.J.; Seo, D.M.; Wang, L.; Beecham, G.W.; Liu, Z.J.; Vazquez-Padron, R.I.; Dong, C.; Hare, J.M.; Kapiloff, M.S.; Bishopric, N.H.; et al. Inflammation, stem cells and atherosclerosis genetics. Curr. Opin. Mol. Ther 2010, 12, 712–723. [Google Scholar]
- Dauwe, D.F.; Janssens, S.P. Stem cell therapy for the treatment of myocardial infarction. Curr. Pharm. Des 2011, 17, 3328–3340. [Google Scholar]
- Strauer, B.E.; Steinhoff, G. 10 years of intracoronary and intramyocardial bone marrow stem cell therapy of the heart: from the methodological origin to clinical practice. J. Am. Coll. Cardiol 2011, 58, 1095–1104. [Google Scholar]
- Feng, Y.; Yang, S.H.; Xiao, B.J.; Xu, W.H.; Ye, S.N.; Xia, T.; Zheng, D.; Liu, X.Z.; Liao, Y.F. Decreased in the number and function of circulation endothelial progenitor cells in patients with avascular necrosis of the femoral head. Bone 2010, 46, 32–40. [Google Scholar]







© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tonini, C.L.; Campagnaro, B.P.; Louro, L.P.S.; Pereira, T.M.C.; Vasquez, E.C.; Meyrelles, S.S. Effects of Aging and Hypercholesterolemia on Oxidative Stress and DNA Damage in Bone Marrow Mononuclear Cells in Apolipoprotein E-deficient Mice. Int. J. Mol. Sci. 2013, 14, 3325-3342. https://doi.org/10.3390/ijms14023325
Tonini CL, Campagnaro BP, Louro LPS, Pereira TMC, Vasquez EC, Meyrelles SS. Effects of Aging and Hypercholesterolemia on Oxidative Stress and DNA Damage in Bone Marrow Mononuclear Cells in Apolipoprotein E-deficient Mice. International Journal of Molecular Sciences. 2013; 14(2):3325-3342. https://doi.org/10.3390/ijms14023325
Chicago/Turabian StyleTonini, Clarissa L., Bianca P. Campagnaro, Lis P. S. Louro, Thiago M. C. Pereira, Elisardo C. Vasquez, and Silvana S. Meyrelles. 2013. "Effects of Aging and Hypercholesterolemia on Oxidative Stress and DNA Damage in Bone Marrow Mononuclear Cells in Apolipoprotein E-deficient Mice" International Journal of Molecular Sciences 14, no. 2: 3325-3342. https://doi.org/10.3390/ijms14023325
APA StyleTonini, C. L., Campagnaro, B. P., Louro, L. P. S., Pereira, T. M. C., Vasquez, E. C., & Meyrelles, S. S. (2013). Effects of Aging and Hypercholesterolemia on Oxidative Stress and DNA Damage in Bone Marrow Mononuclear Cells in Apolipoprotein E-deficient Mice. International Journal of Molecular Sciences, 14(2), 3325-3342. https://doi.org/10.3390/ijms14023325