A Review of the “Omics” Approach to Biomarkers of Oxidative Stress in Oryza sativa



Abstract
:1. Introduction
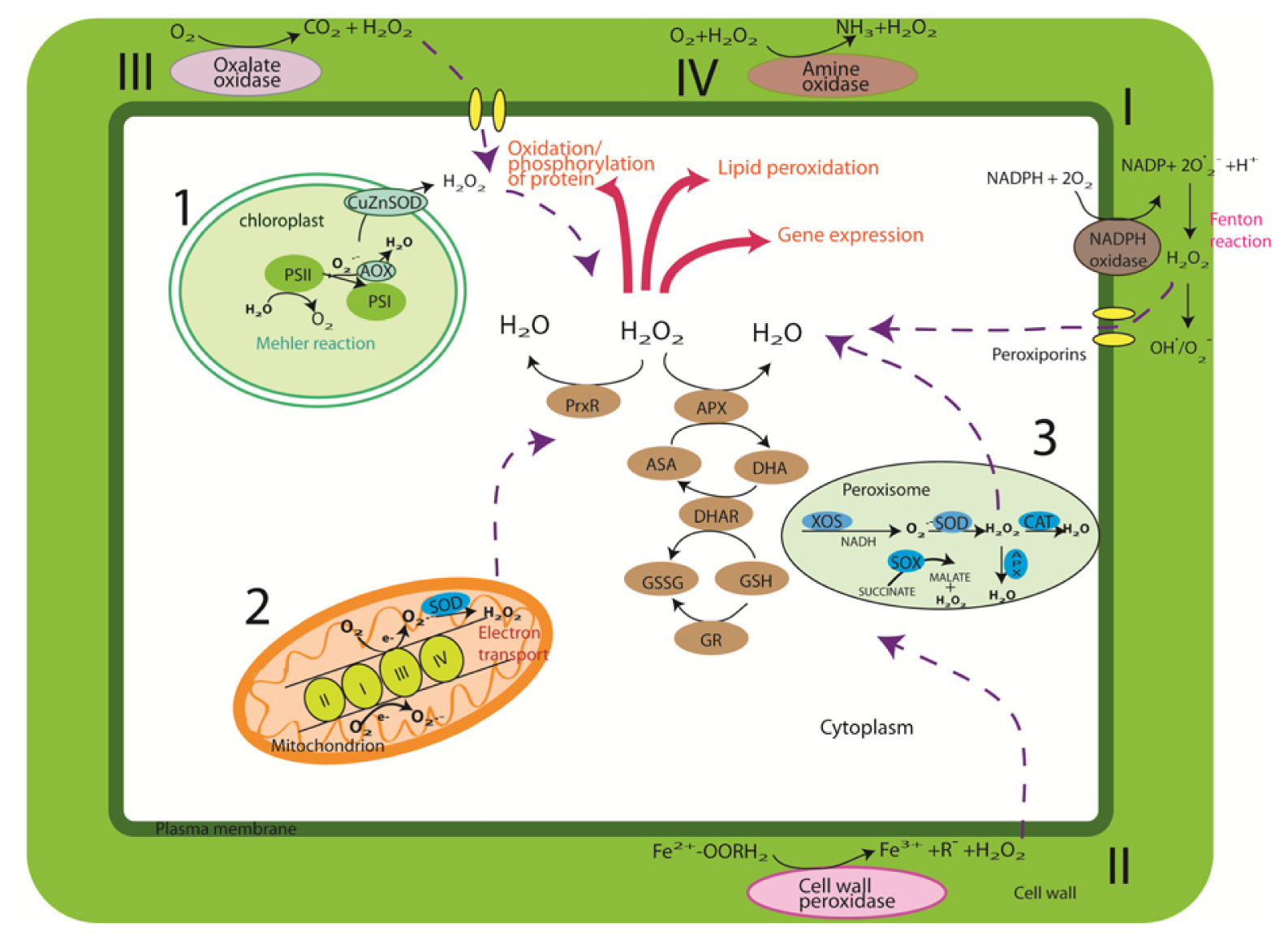
2. Cellular Sources and Regulation of ROS
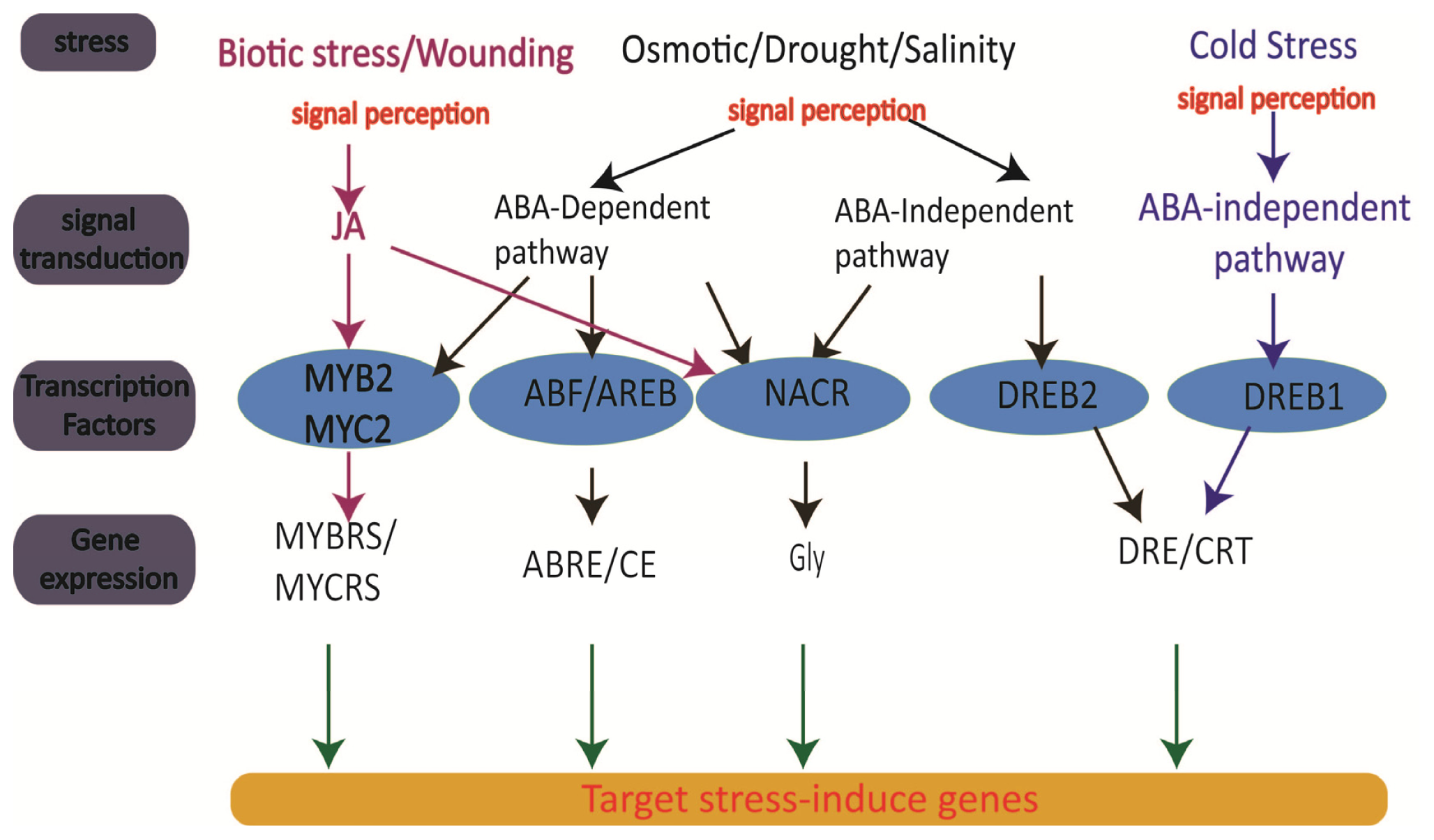
3. Genomics of Rice
4. Rice Proteomics
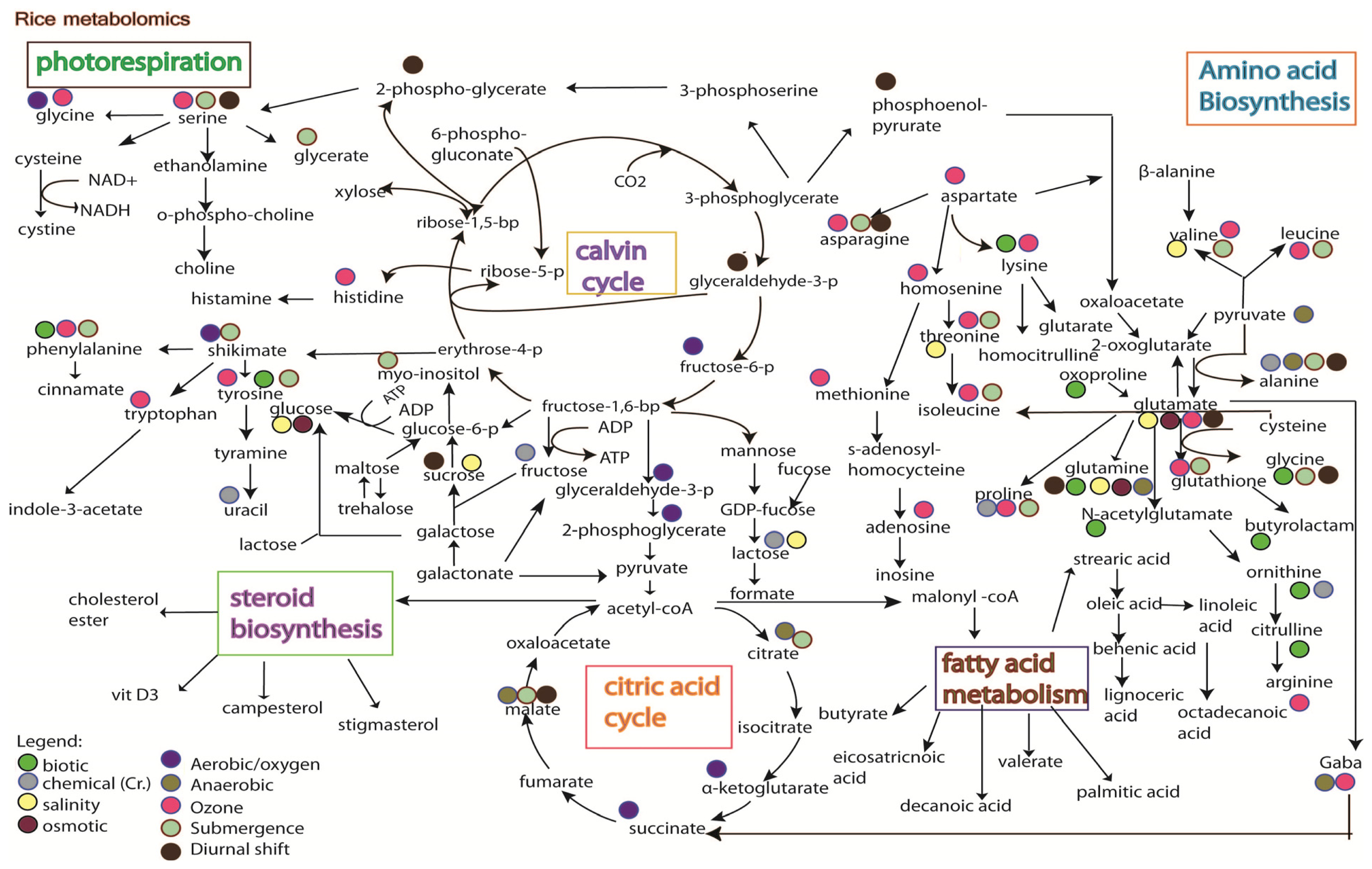
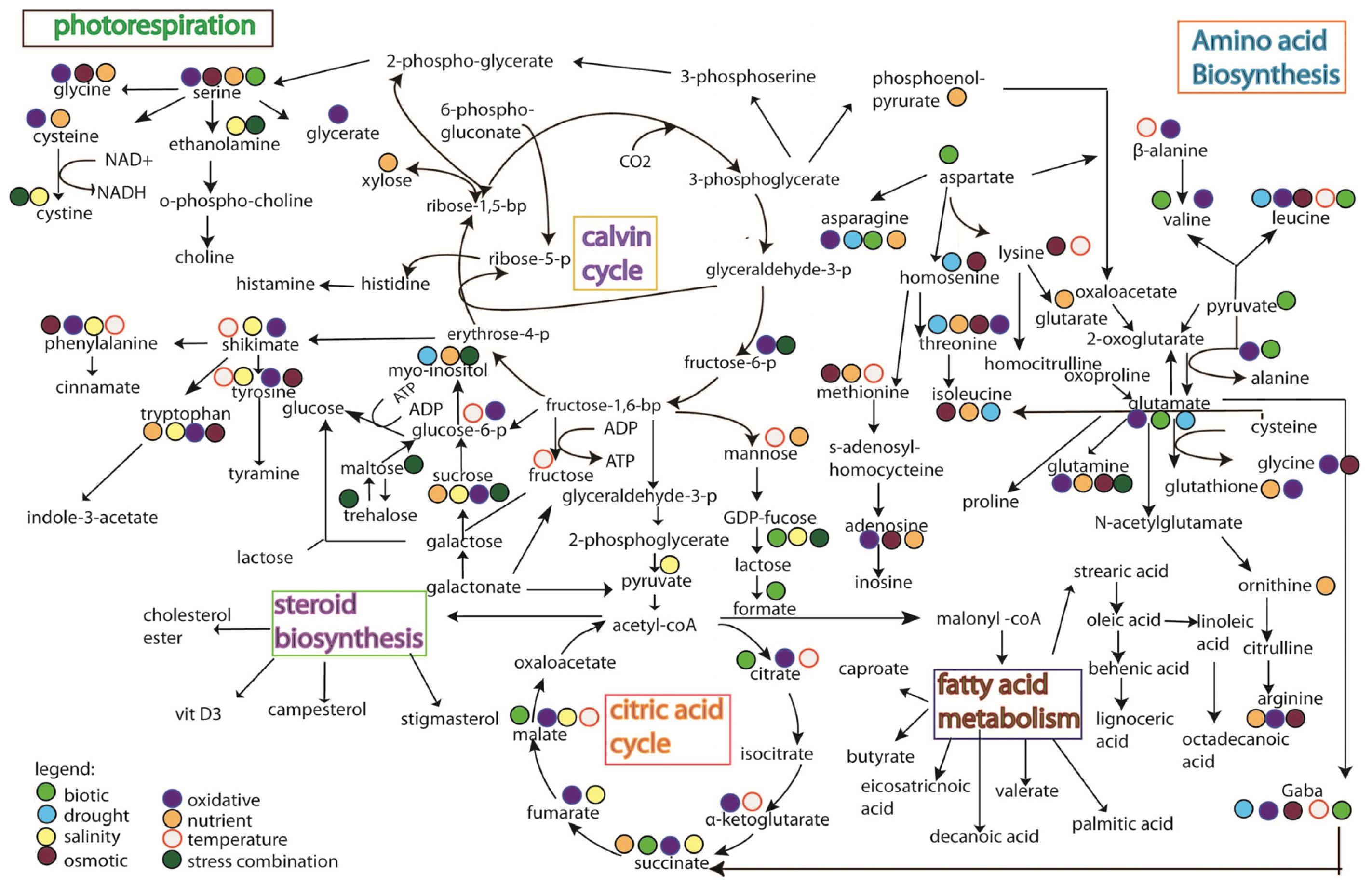
5. Rice Metabolomics
6. Summary of Omic’s Data Set and the Challenge of Integrating Multi-Omic Data Sets
- (1)
- Weak biomarker characterization and validation strategies;
- (2)
- Limitation of the analytical techniques used. As shown in Figures 3 and 4, it is hard to conclude whether the differences in metabolites identified from the same stress is due to species-specific response, or instead merely reflects the variation between analytical tools, e.g., the use of GC versus the use of NMR;
- (3)
- Difficulty in obtaining highly sensitive and specific biomarkers.
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Krishnaiah, K.; Varma, N.G. Changing insect pest scenario in the rice ecosystem—A national perspective. Rice Knowl. Manag. Portal 2011, 1–28. [Google Scholar]
- Datta, S.K. Rice improvement through application of biotechnological tools. Rice Knowl. Manag. Portal 2011, 1–35. [Google Scholar]
- Redona, E.D. Rice biotechnology for developing countries in Asia. Agric. Biotechnol. 2004, 221–230. [Google Scholar]
- Newton, A.C.; Johnson, S.N.; Gregory, P.J. Implications of climate change for diseases, crop yields and food security. Euphytica 2011, 179, 3–18. [Google Scholar]
- De Azevedo Neto, A.D.; Prisco, J.T.; Enéas-Filho, J.; Rolim Medeiros, J.V.; Gomes-Filho, E. Hydrogen peroxide pre-treatment induces salt-stress acclimation in maize plants. J. Plant Physiol 2005, 162, 1114–1122. [Google Scholar]
- Cheeseman, J.M. Hydrogen peroxide and plant stress: A challenge relationship. Plant Stress 2007, 1, 4–15. [Google Scholar]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol 2006, 141, 312–322. [Google Scholar]
- Gadjev, I.; Stone, J.M.; Gechev, T.S. Programmed Cell Death in Plants: New Insights into Redox Regulation and the Role of Hydrogen Peroxide. In International Review of Cell and Molecular Biology; Kwang, W.J., Ed.; Academic Press: Waltham, MA, USA, 2008; Volume 270, pp. 87–144. [Google Scholar]
- Neill, S.J.; Desikan, R.; Clarke, A.; Hurst, R.D.; Hancock, J.T. Hydrogen peroxide and nitric oxide as signalling molecules in plants. J. Exp. Bot 2002, 53, 1237–1247. [Google Scholar]
- Bolwell, G.P.; Wojtaszek, P. Mechanisms for the generation of reactive oxygen species in plant defence—A broad perspective. Physiol. Mol. Plant Pathol 1997, 51, 347–366. [Google Scholar]
- Orozco-Cárdenas, M.L.; Narváez-Vásquez, J.; Ryan, C.A. Hydrogen peroxide acts as a second messenger for the induction of defense genes in tomato plants in response to wounding, systemin, and methyl jasmonate. Plant Cell Online 2001, 13, 179–191. [Google Scholar]
- Desikan, R.; Clarke, A.; Hancock, J.T.; Neill, S.J. H2O2 activates a MAP kinase-like enzyme in Arabidopsis thaliana suspension cultures. J. Exp. Bot 1999, 50, 1863–1866. [Google Scholar]
- Desikan, R.; Cheung, M.K.; Clarke, A.; Golding, S.; Sagi, M.; Fluhr, R.; Rock, C.; Hancock, J.; Neill, S. Hydrogen peroxide is a common signal for darkness-and ABA-induced stomatal closure in Pisum sativum. Funct. Plant Biol 2004, 31, 913–920. [Google Scholar]
- Desikan, R.; Reynolds, A.; Hancock, J.T.; Neill, S.J. Harpin and hydrogen peroxide both initiate programmed cell death but have differential effects on defence gene expression in Arabidopsis suspension cultures. Biochem. J 1998, 330, 115–120. [Google Scholar]
- Aihong, L.; Wang, Y.; Tang, J.; Xue, P.; Li, C.; Liu, L.; Hu, B.; Yang, F.; Loake, G.J.; Chu, C. Nitric oxide and protein s-nitrosylation are integral to hydrogen peroxide induced leaf cell death in rice. Plant Physiol 2012, 158, 451–464. [Google Scholar]
- Białkowski, K.; Olinski, R. Oxidative damage to plant DNA in relation to growth conditions. Acta Biochim. Polonica 1999, 46, 43–49. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Britt, A.B. DNA damage and repair in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol 1996, 47, 75–100. [Google Scholar]
- Moller, I.M.; Jensen, P.E.; Hansson, A. Oxidative modifications to cellular components in plants. Annu. Rev. Plant Biol 2007, 58, 459–481. [Google Scholar]
- Ghezzi, P.; Bonetto, V. Redox proteomics: Identification of oxidatively modified proteins. Proteomics 2003, 3, 1145–1153. [Google Scholar]
- Kukreja, S.; Nandwal, A.; Kumar, N.; Sharma, S.; Unvi, V.; Sharma, P. Plant water status, H2O2 scavenging enzymes, ethylene evolution and membrane integrity of Cicer arietinum roots as affected by salinity. Biol. Plant 2005, 49, 305–308. [Google Scholar]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol 2004, 55, 373–399. [Google Scholar]
- Hung, W.-C.; Huang, D.-D.; Chien, P.-S.; Yeh, C.-M.; Chen, P.-Y.; Chi, W.-C.; Huang, H.-J. Protein tyrosine dephosphorylation during copper-induced cell death in rice roots. Chemosphere 2007, 69, 55–62. [Google Scholar]
- Keller, R.J.; Halmes, N.C.; Hinson, J.A.; Pumford, N.R. Immunochemical detection of oxidized proteins. Chem. Res. Toxicol 1993, 6, 430–433. [Google Scholar]
- Nguyen, A.; Donaldson, R. Metal-catalyzed oxidation induces carbonylation of peroxisomal proteins and loss of enzymatic activities. Arch. Biochem. Biophys 2005, 439, 25–31. [Google Scholar]
- Vanacker, H.; Sandalio, L.; Jimenez, A.; Palma, J.; Corpas, F.; Meseguer, V.; Gomez, M.; Sevilla, F.; Leterrier, M.; Foyer, C.; et al. Roles for redox regulation in leaf senescence of pea plants grown on different sources of nitrogen nutrition. J. Exp. Bot 2006, 57, 1735–1745. [Google Scholar]
- Peng, S.; Huang, J.; Sheehy, J.E.; Laza, R.C.; Visperas, R.M.; Zhong, X.; Centeno, G.S.; Khush, G.S.; Cassman, K.G. Rice yields decline with higher night temperature from global warming. Proc. Natl. Acad. Sci. USA 2004, 101, 9971–9975. [Google Scholar]
- Queval, G.; Hager, J.; Gakiere, B.; Noctor, G. Why are literature data for H2O2 contents so variable? A discussion of potential difficulties in the quantitative assay of leaf extracts. J. Exp. Bot 2008, 59, 135–146. [Google Scholar]
- Foyer, C.H. Redox sensing and signalling associated with reactive oxygen in chloroplasts, peroxisomes and mitochondria. Physiol. Plant 2003, 119, 355–364. [Google Scholar]
- Neill, S.; Desikan, R.; Hancock, J. Hydrogen peroxide signalling. Curr. Opin. Plant Biol 2002, 5, 388–395. [Google Scholar]
- Moller, I.M. Plant mitochondria and oxidative stress: Electron transport, NADPH turnover, and metabolism of reactive oxygen species. Annu. Rev. Plant Biol 2001, 52, 561–591. [Google Scholar]
- Asada, K. Production and scavenging of reactive oxygen species in chloroplasts and their functions. Plant Physiol 2006, 141, 391–396. [Google Scholar]
- Sano, S.; Tao, S.; Endo, Y.; Inaba, T.; Hossain, M.A.; Miyake, C.; Matsuo, M.; Aoki, H.; Asada, K.; Saito, K. Purification and cDNA cloning of chloroplastic monodehydroascorbate reductase from spinach. Biosci. Biotechnol. Biochem 2005, 69, 762–772. [Google Scholar]
- Del Rio, L.A.; Sandalio, L.M.; Corpas, F.J.; Palma, J.M.; Barroso, J.B. Reactive oxygen species and reactive nitrogen species in peroxisomes. Production, scavenging, and role in cell signaling. Plant Physiol 2006, 141, 330–335. [Google Scholar]
- Mano, S.; Nishimura, M. Plant Peroxisomes. In Vitamins and Hormones; Gerald, L., Ed.; Academic Press: Waltham, MA, USA, 2005; Volume 72, pp. 111–154. [Google Scholar]
- Noctor, G.; de Paepe, R.; Foyer, C.H. Mitochondrial redox biology and homeostasis in plants. Trends Plant Sci 2007, 12, 125–134. [Google Scholar]
- Rhoads, D.M.; Umbach, A.L.; Subbaiah, C.C.; Siedow, J.N. Mitochondrial reactive oxygen species. Contribution to oxidative stress and interorganellar signaling. Plant Physiol 2006, 141, 357–366. [Google Scholar]
- Lherminier, J.; Elmayan, T.; Fromentin, J.R.M.; Elaraqui, K.T.; Vesa, S.; Morel, J.; Verrier, J.-L.; Cailleteau, B.; Blein, J.-P.; Simon-Plas, F.O. NADPH oxidase-mediated reactive oxygen species production: Subcellular localization and reassessment of its role in plant defense. Mol. Plant Microbe Interact 2009, 22, 868–881. [Google Scholar]
- Kawasaki, S.; Borchert, C.; Deyholos, M.; Wang, H.; Brazille, S.; Kawai, K.; Galbraith, D.; Bohnert, H.J. Gene expression profiles during the initial phase of salt stress in rice. Plant Cell 2001, 13, 889–906. [Google Scholar]
- Gorantla, M.; Babu, P.; Reddy, L.V.; Reddy, A.; Wusirika, R.; Bennetzen, J.L.; Reddy, A.R. Identification of stress-responsive genes in an indica rice (Oryza sativa L.) using ESTs generated from drought-stressed seedlings. J. Exp. Bot 2007, 58, 253–265. [Google Scholar]
- Yamakawa, H.; Hirose, T.; Kuroda, M.; Yamaguchi, T. Comprehensive expression profiling of rice grain filling-related genes under high temperature using DNA microarray. Plant Physiol 2007, 144, 258–277. [Google Scholar]
- Cheng, C.; Yun, K.-Y.; Ressom, H.; Mohanty, B.; Bajic, V.; Jia, Y.; Yun, S.; de los Reyes, B. An early response regulatory cluster induced by low temperature and hydrogen peroxide in seedlings of chilling-tolerant japonica rice. BMC Genomics 2007, 8, 175. [Google Scholar]
- Jung, K.-H.; Dardick, C.; Bartley, L.E.; Cao, P.; Phetsom, J.; Canlas, P.; Seo, Y.-S.; Shultz, M.; Ouyang, S.; Yuan, Q.; et al. Refinement of light-responsive transcript lists using rice oligonucleotide arrays: Evaluation of gene-redundancy. PLoS One 2008, 3, e3337. [Google Scholar]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K.; Ito, Y.; Yoshiwara, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol 2003, 133, 1755–1767. [Google Scholar]
- Nakashima, K.; Tran, L.S.P.; van Nguyen, D.; Fujita, M.; Maruyama, K.; Todaka, D.; Ito, Y.; Hayashi, N.; Shinozaki, K.; Yamaguchi‚, S.K. Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J. 2007, 51, 617–630. [Google Scholar]
- Quan, R.; Hu, S.; Zhang, Z.; Zhang, H.; Zhang, Z.; Huang, R. Overexpression of an ERF transcription factor TSRF1 improves rice drought tolerance. Plant Biotechnol. J 2010, 8, 476–488. [Google Scholar]
- Mukherjee, K.; Choudhury, A.; Gupta, B.; Gupta, S.; Sengupta, D. An ABRE-binding factor, OSBZ8, is highly expressed in salt tolerant cultivars than in salt sensitive cultivars of indica rice. BMC Plant Biol 2006, 6, 18. [Google Scholar]
- Dubouzet, J.G.; Sakuma, Y.; Ito, Y.; Kasuga, M.; Dubouzet, E.G.; Miura, S.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J 2003, 33, 751–763. [Google Scholar]
- Prashanth, S.; Sadhasivam, V.; Parida, A. Over expression of cytosolic copper/zinc superoxide dismutase from a mangrove plant Avicennia marina in indica Rice var Pusa Basmati-1 confers abiotic stress tolerance. Transgenic Res 2008, 17, 281–291. [Google Scholar]
- Wang, F.-Z.; Wang, Q.-B.; Kwon, S.-Y.; Kwak, S.-S.; Su, W.-A. Enhanced drought tolerance of transgenic rice plants expressing a pea manganese superoxide dismutase. J. Plant Physiol 2005, 162, 465–472. [Google Scholar]
- Tanaka, Y.; Hibino, T.; Hayashi, Y.; Tanaka, A.; Kishitani, S.; Takabe, T.; Yokota, S. Salt tolerance of transgenic rice overexpressing yeast mitochondrial Mn-SOD in chloroplasts. Plant Sci 1999, 148, 131–138. [Google Scholar]
- Nagamiya, K.; Motohashi, T.; Nakao, K.; Prodhan, S.; Hattori, E.; Hirose, S.; Ozawa, K.; Ohkawa, Y.; Takabe, T.; Takabe, T.; et al. Enhancement of salt tolerance in transgenic rice expressing an Escherichia coli catalase gene, kat E. Plant Biotechnol. Rep 2007, 1, 49–55. [Google Scholar]
- Matsumura, T.; Tabayashi, N.; Kamagata, Y.; Souma, C.; Saruyama, H. Wheat catalase expressed in transgenic rice can improve tolerance against low temperature stress. Physiol. Plant 2002, 116, 317–327. [Google Scholar]
- Zhao, F.; Zhang, H. Salt and paraquat stress tolerance results from co-expression of the Suaeda salsa glutathione S-transferase and catalase in transgenic rice. Plant Cell Tissue Organ Culture 2006, 86, 349–358. [Google Scholar]
- Bonifacio, A.; Martins, M.O.; Ribeiro, C.W.; Fontenele, A.V.; Carvalho, F.E.L.; Margis-Pinheiro, M.; Silveira, J.A.G. Role of peroxidases in the compensation of cytosolic ascorbate peroxidase knockdown in rice plants under abiotic stress. Plant Cell Environ 2011, 34, 1705–1722. [Google Scholar]
- Sato, Y.; Masuta, Y.; Saito, K.; Murayama, S.; Ozawa, K. Enhanced chilling tolerance at the booting stage in rice by transgenic overexpression of the ascorbate peroxidase gene, OsAPXa. Plant Cell Rep 2011, 30, 399–406. [Google Scholar]
- Kouril, R.; Lazar, D.; Lee, H.; Jo, J.; Naus, J. Moderately elevated temperature eliminates resistance of rice plants with enhanced expression of glutathione reductase to intensive photooxidative stress. Photosynthetica 2003, 41, 571–578. [Google Scholar]
- Takesawa, T.; Ito, M.; Kanzaki, H.; Kameya, N.; Nakamura, I. Over-expression of zeta glutathione S-transferase in transgenic rice enhances germination and growth at low temperature. Mol. Breed 2002, 9, 93–101. [Google Scholar]
- Hu, T.; Qv, X.; Xiao, G.; Huang, X. Enhanced tolerance to herbicide of rice plants by over-expression of a glutathione S-transferase. Mol. Breed 2009, 24, 409–418. [Google Scholar]
- Olsen, A.N.; Ernst, H.; Leggio, L.L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci 2005, 10, 79–87. [Google Scholar]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar]
- Ohnishi, T.; Sugahara, S.; Yamada, T.; Kikuchi, K.; Yoshiba, Y.; Hirano, H.; Tsutsumi, N. OsNAC6, a member of the NAC gene family, is induced by various stresses in rice. Genes Genet. Syst 2005, 80, 135. [Google Scholar]
- Ren, Z.-H.; Gao, J.-P.; Li, L.-G.; Cai, X.-L.; Huang, W.; Chao, D.-Y.; Zhu, M.-Z.; Wang, Z.-Y.; Luan, S.; Lin, H.-X. A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat. Genet 2005, 37, 1141–1146. [Google Scholar]
- Lu, C.-A.; Ho, T.-H.D.; Ho, S.-L.; Yu, S.-M. Three novel MYB proteins with one DNA binding repeat rediate sugar and hormone regulation of α-amylase gene expression. Plant Cell Online 2002, 14, 1963–1980. [Google Scholar]
- Su, C.-F.; Wang, Y.-C.; Hsieh, T.-H.; Lu, C.-A.; Tseng, T.-H.; Yu, S.-M. A novel MYBS3-dependent pathway confers cold tolerance in rice. Plant Physiol 2010, 153, 145–158. [Google Scholar]
- Chen, Y.; Yang, X.; He, K.; Liu, M.; Li, J.; Gao, Z.; Lin, Z.; Zhang, Y.; Wang, X.; Qiu, X.; et al. The MYB transcription factor superfamily of Arabidopsis: Expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol 2006, 60, 107–124. [Google Scholar]
- Gadjev, I.; Vanderauwera, S.; Gechev, T.S.; Laloi, C.; Minkov, I.N.; Shulaev, V.; Apel, K.; Inzé, D.; Mittler, R.; van Breusegem, F. Transcriptomic footprints disclose specificity of reactive oxygen species signaling in Arabidopsis. Plant Physiol 2006, 141, 436–445. [Google Scholar]
- Cho, K.; Shibato, J.; Agrawal, G.K.; Jung, Y.-H.; Kubo, A.; Jwa, N.-S.; Tamogami, S.; Satoh, K.; Kikuchi, S.; Higashi, T.; et al. Integrated transcriptomics, proteomics, and metabolomics analyses to survey ozone responses in the leaves of rice seedling. J. Proteome Res 2008, 7, 2980–2998. [Google Scholar]
- Xu, K.; Xu, X.; Fukao, T.; Canlas, P.; Maghirang-Rodriguez, R.; Heuer, S.; Ismail, A.M.; Bailey-Serres, J.; Ronald, P.C.; Mackill, D.J. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 2006, 442, 705–708. [Google Scholar]
- Liu, F.; Xu, W.; Wei, Q.; Zhang, Z.; Xing, Z.; Tan, L.; Di, C.; Yao, D.; Wang, C.; Tan, Y.; et al. Gene expression profiles deciphering rice phenotypic variation between Nipponbare (Japonica) and 93-11 (Indica) during oxidative stress. PLoS One 2010, 5, e8632. [Google Scholar]
- Komatsu, S. Rice Proteomics: A Step toward Functional Analysis of the Rice Genome. In Rice Functional Genomics; Springer: New York, NY, USA, 2007; pp. 61–89. [Google Scholar]
- Schroeder, J.I.; Kwak, J.M.; Allen, G.J. Guard cell abscisic acid signalling and engineering drought hardiness in plants. Nature 2001, 410, 327–330. [Google Scholar]
- Fukuda, M.; Islam, N.; Woo, S.-H.; Yamagishi, A.; Takaoka, M.; Hirano, H. Assessing matrix assisted laser desorption/ionization-time of flight-mass spectrometry as a means of rapid embryo protein identification in rice. Electrophoresis 2003, 24, 1319–1329. [Google Scholar]
- Chen, X.; Wang, Y.; Li, J.; Jiang, A.; Cheng, Y.; Zhang, W. Mitochondrial proteome during salt stress-induced programmed cell death in rice. Plant Physiol. Biochem 2009, 47, 407–415. [Google Scholar]
- Yan, S.; Tang, Z.; Su, W.; Sun, W. Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics 2005, 5, 235–244. [Google Scholar]
- Lee, D.-G.; Ahsan, N.; Lee, S.-H.; Lee, J.J.; Bahk, J.D.; Kang, K.Y.; Lee, B.-H. Chilling stress-induced proteomic changes in rice roots. J. Plant Physiol 2009, 166, 1–11. [Google Scholar]
- Salekdeh, G.H.; Siopongco, J.; Wade, L.J.; Ghareyazie, B.; Bennett, J. Proteomic analysis of rice leaves during drought stress and recovery. Proteomics 2002, 2, 1131–1145. [Google Scholar]
- Liu, J.-X.; Bennett, J. Reversible and irreversible drought-induced changes in the anther proteome of rice (Oryza sativa L.) genotypes IR64 and moroberekan. Mol. Plant 2011, 4, 59–69. [Google Scholar]
- Rao, S.R.; Ford, K.L.; Cassin, A.M.; Roessner, U.; Patterson, J.H.; Bacic, A. Proteomic and metabolic profiling of rice suspension culture cells as a model to study abscisic acid signaling response pathways in plants. J. Proteome Res 2010, 9, 6623–6634. [Google Scholar]
- Cai, L.; Tu, B.P. Driving the cell cycle through metabolism. Annu. Rev. Cell Dev. Biol 2012, 28, 59–87. [Google Scholar]
- Cuddihy, S.L.; Baty, J.W.; Brown, K.K.; Winterbourn, C.C.; Hampton, M.B. Proteomic detection of oxidized and reduced thiol proteins in cultured cells. Methods Mol Biol 2008, 519, 363–375. [Google Scholar]
- Baty, J.W.; Hampton, M.B.; Winterbourn, C.C. Detection of oxidant sensitive thiol proteins by fluorescence labeling and two-dimensional electrophoresis. Proteomics 2002, 2, 1261–1266. [Google Scholar]
- Fu, C.; Hu, J.; Liu, T.; Ago, T.; Sadoshima, J.; Li, H. Quantitative analysis of redox-sensitive proteome with DIGE and ICAT. J. Proteome Res 2008, 7, 3789–3802. [Google Scholar]
- Koller, A.; Washburn, M.P.; Lange, B.M.; Andon, N.L.; Deciu, C.; Haynes, P.A.; Hays, L.; Schieltz, D.; Ulaszek, R.; Wei, J.; et al. Proteomic survey of metabolic pathways in rice. Proc. Natl. Acad. Sci. USA 2002, 99, 11969–11974. [Google Scholar]
- Griffin, T.J.; Sherman, J.; Aebersold, R. Quantitative Proteomics (ICAT™); John Wiley & Sons Ltd: Hoboken, NJ, USA, 2001. [Google Scholar]
- Ali, G.M.; Komatsu, S. Proteomic analysis of rice leaf sheath during drought stress. J. Proteome Res 2006, 5, 396–403. [Google Scholar]
- Abbasi, F.M.; Komatsu, S. A proteomic approach to analyze salt-responsive proteins in rice leaf sheath. Proteomics 2004, 4, 2072–2081. [Google Scholar]
- Parker, R.; Flowers, T.J.; Moore, A.L.; Harpham, N.V.J. An accurate and reproducible method for proteome profiling of the effects of salt stress in the rice leaf lamina. J. Exp. Bot 2006, 57, 1109–1118. [Google Scholar]
- Zang, X.; Komatsu, S. A proteomics approach for identifying osmotic-stress-related proteins in rice. Phytochemistry 2007, 68, 426–437. [Google Scholar]
- Xiong, J.-H.; Fu, B.-Y.; Xu, H.-X.; LI, Y.-S. Proteomic analysis of PEG-simulated drought stress-responsive proteins of rice leaves using a pyramiding rice line at the seedling stage. Bot. Studies 2010, 51, 137–145. [Google Scholar]
- Lee, D.-G.; Ahsan, N.; Lee, S.-H.; Kang, K.Y.; Bahk, J.D.; Lee, I.-J.; Lee, B.-H. A proteomic approach in analyzing heat-responsive proteins in rice leaves. Proteomics 2007, 7, 3369–3383. [Google Scholar]
- Cui, S.; Huang, F.; Wang, J.; Ma, X.; Cheng, Y.; Liu, J. A proteomic analysis of cold stress responses in rice seedlings. Proteomics 2005, 5, 3162–3172. [Google Scholar]
- Makoto, H.; Setsuko, K. Proteomic analysis of rice seedlings during cold stress. Proteomics 2007, 7, 1293–1302. [Google Scholar]
- Lee, D.-G.; Ahsan, N.; Lee, S.-H.; Kang, K.Y.; Lee, J.J.; Lee, B.H. An approach to identify cold-induced low-abundant proteins in rice leaf. C. R. Biol 2007, 330, 215–225. [Google Scholar]
- Chen, S.; Zeng, F.; Wu, F.; Ma, W.; Zhang, G. Proteomic analysis of nitrogen stress-responsive proteins in two rice cultivars differing in N utilization efficiency. J. Integr. OMICS 2011, 1, 22. [Google Scholar]
- Lee, K.; Bae, D.W.; Kim, S.H.; Han, H.J.; Liu, X.; Park, H.C.; Lim, C.O.; Lee, S.Y.; Chung, W.S. Comparative proteomic analysis of the short-term responses of rice roots and leaves to cadmium. J. Plant Physiol 2010, 167, 161–168. [Google Scholar]
- Royuela, M.; Gonzalez, A.; Gonzalez, E.M.; Arrese-Igor, C.; Aparicio-Tejo, P.M.; Gonzalez-Murua, C. Physiological consequences of continuous, sublethal imazethapyr supply to pea plants. J. Plant Physiol 2000, 157, 345–354. [Google Scholar]
- Hollywood, K.; Brison, D.R.; Goodacre, R. Metabolomics: Current technologies and future trends. Proteomics 2006, 6, 4716–4723. [Google Scholar]
- Okazaki, Y.; Saito, K. Recent advances of metabolomics in plant biotechnology. Plant Biotechnol. Rep 2012, 6, 1–15. [Google Scholar]
- Johnson, H.E.; Broadhurst, D.; Goodacre, R.; Smith, A.R. Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 2003, 62, 919–928. [Google Scholar]
- Hirai, M.Y.; Yano, M.; Goodenowe, D.B.; Kanaya, S.; Kimura, T.; Awazuhara, M.; Arita, M.; Fujiwara, T.; Saito, K. Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2004, 101, 10205–10210. [Google Scholar]
- Kaplan, F.; Kopka, J.; Haskell, D.W.; Zhao, W.; Schiller, K.C.; Gatzke, N.; Sung, D.Y.; Guy, C.L. Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiol 2004, 136, 4159–4168. [Google Scholar]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomic analysis of plants. Nat. Protoc 2010, 5, 536–549. [Google Scholar]
- Shu, X.-L.; Frank, T.; Shu, Q.-Y.; Engel, K.-H. Metabolite profiling of germinating rice seeds. J. Agric. Food Chem 2008, 56, 11612–11620. [Google Scholar]
- Wakasa, K.; Hasegawa, H.; Nemoto, H.; Matsuda, F.; Miyazawa, H.; Tozawa, Y.; Morino, K.; Komatsu, A.; Yamada, T.; Terakawa, T.; et al. High-level tryptophan accumulation in seeds of transgenic rice and its limited effects on agronomic traits and seed metabolite profile. J. Exp. Bot 2006, 57, 3069–3078. [Google Scholar]
- Tarpley, L.; Duran, A.; Kebrom, T.; Sumner, L. Biomarker metabolites capturing the metabolite variance present in a rice plant developmental period. BMC Plant Biol 2005, 5, 8. [Google Scholar]
- Kusano, M.; Fukushima, A.; Kobayashi, M.; Hayashi, N.; Jonsson, P.; Moritz, T.; Ebana, K.; Saito, K. Application of a metabolomic method combining one-dimensional and two-dimensional gas chromatography-time-of-flight/mass spectrometry to metabolic phenotyping of natural variants in rice. J. Chromatogr. B 2007, 855, 71–79. [Google Scholar]
- Fumagalli, E.; Baldoni, E.; Abbruscato, P.; Piffanelli, P.; Genga, A.; Lamanna, R.; Consonni, R. NMR techniques coupled with multivariate statistical analysis: Tools to analyse Oryza sativa metabolic content under stress conditions. J. Agron. Crop Sci 2009, 195, 77–88. [Google Scholar]
- Sana, T. Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 2010, 6, 451–465. [Google Scholar]
- Dubey, S.; Misra, P.; Dwivedi, S.; Chatterjee, S.; Bag, S.; Mantri, S.; Asif, M.; Rai, A.; Kumar, S.; Shri, M.; et al. Transcriptomic and metabolomic shifts in rice roots in response to Cr (VI) stress. BMC Genomics 2010, 11, 648. [Google Scholar]
- Fan, T.W.M. In vivo and in vitro metabolomic analysis of anaerobic rice coleoptiles revealed unexpected pathways. Rus. J. Plant Physiol 2003, 50, 787–793. [Google Scholar]
- Narsai, R.; Howell, K.A.; Carroll, A.; Ivanova, A.; Millar, A.H.; Whelan, J. Defining core metabolic and transcriptomic responses to oxygen availability in rice embryos and young seedlings. Plant Physiol 2009, 151, 306–322. [Google Scholar]
- Barding, G.A.; Béni, S.; Fukao, T.; Bailey-Serres, J.; Larive, C.K. Comparison of GC-MS and NMR for metabolite profiling of rice subjected to submergence stress. J. Proteome Res 2012, 12, 898–909. [Google Scholar]
- Ishikawa, T.; Takahara, K.; Hirabayashi, T.; Matsumura, H.; Fujisawa, S.; Terauchi, R.; Uchimiya, H.; Kawai-Yamada, M. Metabolome analysis of response to oxidative stress in rice suspension cells overexpressing cell death suppressor bax inhibitor-1. Plant Cell Physiol 2010, 51, 9–20. [Google Scholar]
- Sato, S.; Arita, M.; Soga, T.; Nishioka, T.; Tomita, M. Time-resolved metabolomics reveals metabolic modulation in rice foliage. BMC Syst. Biol 2008, 2, 51. [Google Scholar]
- Kim, J.K.; Bamba, T.; Harada, K.; Fukusaki, E.; Kobayashi, A. Time-course metabolic profiling in Arabidopsis thaliana cell cultures after salt stress treatment. J. Exp. Bot 2007, 58, 415–424. [Google Scholar]
- Charlton, A.; Donarski, J.; Harrison, M.; Jones, S.; Godward, J.; Oehlschlager, S.; Arques, J.; Ambrose, M.; Chinoy, C.; Mullineaux, P.; et al. Responses of the pea (Pisum sativum L.) leaf metabolome to drought stress assessed by nuclear magnetic resonance spectroscopy. Metabolomics 2008, 4, 312–327. [Google Scholar]
- Zulak, K.; Weljie, A.; Vogel, H.; Facchini, P. Quantitative 1H NMR metabolomics reveals extensive metabolic reprogramming of primary and secondary metabolism in elicitor-treated opium poppy cell cultures. BMC Plant Biol 2008, 8, 5. [Google Scholar]
- Lugan, R.; Niogret, M.F.; Leport, L.; Guégan, J.P.; Larher, F.R.; Savouré, A.; Kopka, J.; Bouchereau, A. Metabolome and water homeostasis analysis of Thellungiella salsuginea suggests that dehydration tolerance is a key response to osmotic stress in this halophyte. Plant J 2010, 64, 215–229. [Google Scholar]
- Yamakawa, H.; Hakata, M. Atlas of rice grain filling-related metabolism under high temperature: joint analysis of metabolome and transcriptome demonstrated inhibition of starch accumulation and induction of amino acid accumulation. Plant Cell Physiol 2010, 51, 1599. [Google Scholar]
- Baxter, C.J.; Redestig, H.; Schauer, N.; Repsilber, D.; Patil, K.R.; Nielsen, J.; Selbig, J.; Liu, J.; Fernie, A.R.; Sweetlove, L.J. The metabolic response of heterotrophic Arabidopsis cells to oxidative stress. Plant Physiol 2007, 143, 312–325. [Google Scholar]
- Rizhsky, L.; Liang, H.; Shuman, J.; Shulaev, V.; Davletova, S.; Mittler, R. When defense pathways collide. The response of Arabidopsis to a combination of drought and heat stress. Plant Physiol 2004, 134, 1683–1696. [Google Scholar]
- Wenderoth, I.; Scheibe, R.; von Schaewen, A. Identification of the cysteine residues involved in redox modification of plant plastidic glucose-6-phosphate dehydrogenase. J. Biol. Chem 1997, 272, 26985–26990. [Google Scholar]
- Mailloux, R.J.; Beriault, R.; Lemire, J.; Singh, R.; Chenier, D.R.; Hamel, R.D.; Appanna, V.D. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS One 2007, 2, e690. [Google Scholar]
- Wang, Z.-Q.; Yuan, Y.-Z.; Ou, J.-Q.; Lin, Q.-H.; Zhang, C.-F. Glutamine synthetase and glutamate dehydrogenase contribute differentially to proline accumulation in leaves of wheat (Triticum aestivum) seedlings exposed to different salinity. J. Plant Physiol 2007, 164, 695–701. [Google Scholar]
- Lehmann, M.; Schwarzländer, M.; Obata, T.; Sirikantaramas, S.; Burow, M.; Olsen, C.E.; Tohge, T.; Fricker, M.D.; Møller, B.L.; Fernie, A.R.; et al. The metabolic response of Arabidopsis roots to oxidative stress is distinct from that of heterotrophic cells in culture and highlights a complex relationship between the levels of transcripts, metabolites, and flux. Mol. Plant 2009, 2, 390–406. [Google Scholar]
- Trygg, J.; Holmes, E.; Lundstedt, T.R. Chemometrics in metabonomics. J. Proteome Res 2006, 6, 469–479. [Google Scholar]
- Grimplet, J.; Wheatley, M.D.; Jouira, H.B.; Deluc, L.G.; Cramer, G.R.; Cushman, J.C. Proteomic and selected metabolite analysis of grape berry tissues under well-watered and water-deficit stress conditions. Proteomics 2009, 9, 2503–2528. [Google Scholar]
- Torres-García, W.; Zhang, W.; Runger, G.C.; Johnson, R.H.; Meldrum, D.R. Integrative analysis of transcriptomic and proteomic data of Desulfovibrio vulgaris: A non-linear model to predict abundance of undetected proteins. Bioinformatics 2009, 25, 1905–1914. [Google Scholar]
- Urbanczyk-Wochniak, E.; Luedemann, A.; Kopka, J.; Selbig, J.; Roessner-Tunali, U.; Willmitzer, L.; Fernie, A.R. Parallel analysis of transcript and metabolic profiles: A new approach in systems biology. EMBO Rep 2003, 4, 989–993. [Google Scholar]
- Mochida, K.; Shinozaki, K. Genomics and bioinformatics resources for crop improvement. Plant Cell Physiol 2010, 51, 497–523. [Google Scholar]
- Mochida, K.; Shinozaki, K. Advances in omics and bioinformatics tools for systems analyses of plant functions. Plant Cell Physiol 2011, 52, 2017–2038. [Google Scholar]
- Palsson, B.; Zengler, K. The challenges of integrating multi-omic data sets. Nat. Chem. Biol 2010, 6, 787–789. [Google Scholar]
- Steuer, R. Review: On the analysis and interpretation of correlations in metabolomic data. Brief. Bioinform 2006, 7, 151–158. [Google Scholar]
- Edwards, D.; Batley, J. Plant bioinformatics: from genome to phenome. Trends Biotechnol 2004, 22, 232–237. [Google Scholar]
- Ge, H.; Walhout, A.J.M.; Vidal, M. Integrating “omic” information: A bridge between genomics and systems biology. Trends Genet 2003, 19, 551–560. [Google Scholar]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar]
- Tuncbag, N.; McCallum, S.; Huang, S.S.C.; Fraenkel, E. SteinerNet: A web server for integrating “omic” data to discover hidden components of response pathways. Nucleic Acids Res 2012, 40, W505–W509. [Google Scholar]
- Lysenko, A.; Hindle, M.M.; Taubert, J.; Saqi, M.; Rawlings, C.J. Data integration for plant genomics—exemplars from the integration of Arabidopsis thaliana databases. Brief. Bioinform 2009, 10, 676–693. [Google Scholar]
- Toyoda, T.; Mochizuki, Y.; Player, K.; Heida, N.K.; Naohiko; Sakaki, Y. OmicBrowse: A browser of multidimensional omics annotations. Bioinformatics 2007, 23, 524–526. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Source | Response | Reference |
|---|---|---|---|
| Superoxide dismutase (SOD) | |||
| Cu/Zn SOD | Avicennia marina | Transgenic plants were more tolerant to methyl viologen (MV)-mediated oxidative stress, salinity, and drought stress. | [49] |
| Mn SOD | Pisum sativum | Electrolyte content declined and less injury observed in leaf discs of transgenic plants compared to that observed in wild type plants following treatment with polyethylene glycol (PEG). | [50] |
| Mn SOD | Yeast | Transgenic plants maintained high level of SOD and ascorbate peroxidise activity in chloroplast upon exposure to salt stress, while decrease in SOD activities was observed in wild type plants. | [51] |
| Catalase (CAT) | |||
| CAT KatE | E.coli | Transgenic rice plants showed at least 1.5–2.5 fold increase of CAT upon exposure to salt stress. The transgenic rice can survive for another 14 days compared to wild type plants following treatment with salt stress. | [52] |
| CAT | Triticum aestivum L. | In 5 °C of cold stress, CAT was found 4–15 times higher in transgenic plants compared to that observed in wild type plants with very low levels of H2O2. | [53] |
| CAT | Suaeda salsa | Co-expression of CAT and GST resulted in the increment of SOD and CAT activities following treatment with salt and paraquat stress, while GST activity only increased upon treatment with paraquat stress. H2O2, malondialdehyde, and electrolyte leakage were found to decrease in transgenic rice compared to wild type plants. | [54] |
| Ascorbate peroxidase | |||
| Knockdown OsAPx1 and OsAPx2 | Rice | Compensation of ascorbate peroxidase by other peroxidases, including glutathione peroxidase (GPX). | [55] |
| OsAPXa | Rice | Transgenic plants maintained H2O2 and malondialdehyde (MDA) content when subjected to cold stress. | [56] |
| Silencing APx1/2s | Rice | Upregulation of other peroxidases was observed under salinity, heat, high light, and MV treatment. | [55] |
| Glutathione reductase | |||
| GR | Brassica campestris | High expression of GR improved the protection against photo-bleaching of chlorophyll and photo-oxidative action of MV in thylakoid membranes at 25 °C. | [57] |
| GlutathioneS-transferases | |||
| GST | Rice | Seedlings of the transgenic lines grown under submergence demonstrated enhanced germination and growth rates at low temperature. | [58] |
| OsGSTL1 | Rice | The overexpression lines showed an increase in GST and GPX activities, and a decrease in the level of superoxide was observed. | [59] |
| GST | Suaeda salsa | Salt and paraquat stress tolerance were observed due to GST, CAT, and SOD activity. | [54] |
| Genotype | Treatment | Major result | Reference |
|---|---|---|---|
| Oryza sativa L. cv CT9993 and cv IR62266 | Drought | Signal transduction:Translation elongation factor, actin depolymerizing factor Energy metabolism: ribulose-1, 5-bisphosphate carboxylase/oxygenase (RuBisCo) activase, Triosephosphate isomerase Antioxidant: superoxide dismutase (SOD), GSH-dependent dehydroascorbate reductase, Unknown function: S-like RNase homolog, isoflavone reductase-like protein | [77] |
| Oryza sativa L. Nipponbare Zhonghua 8 | 2–6 days drought | Defense:-superoxide dismutase (SOD), salt-induced protein (SALT), Energy metabolism: chloroplast ATPase, RuBisCO small subunit, RuBisCO large subunit, photosystem II oxygen-evolving complex protein, oxygen-evolving enhancer protein, light harvesting complex chain II Cell structure: Cys peroxiredoxin BAS 1 Signal transduction: actin depolymerizing factor Unknown function: serine hydroxymethyltrasferase I, phosphoglucomutase cytoplasmic | [86] |
| Oryza sativa L. Nipponbare, IR36, Pokkali | Salinity 50 mM NaCl, 24 h | Energy metabolism: photosystem II oxygen-evolving complex protein, oxygen evolving enhancer protein (OEE 2), fructosebisphosphate aldolases Antioxidant-superoxide dismutase (SOD) | [87] |
| Oryza sativa L. IR 4630-22-2- 5-1-3 | Salinity 50 mM NaCl, 7 days | RuBisCo activase (RCA): RuBisCO activase Iron homeostasis: Ferritin, Energy and metabolism: ATP synthase-putative phosphoglycerate kinase, Antioxidant: SOD Metabolism synthesis: S-adenosyl-l-methionine synthetase. Cell cycle: Translation initiation factor 5A | [88] |
| Oryza sativa L. cv. Nipponbare | Salinity 150 mM Nacl, 24 h, 48 h and 72 h | Glycolysis enzyme: Triosephosphate isomerase, Enolase Signal transduction: UDP-glucose pyrophosphorylase (UGPase) Energy generation: Cytochrome c oxidase subunit 6b-1 (COX6b-1), nascent polypeptide-associated complex alpha chain, S-adenosylmethionine synthetase 2 Antioxidants: Glutamate synthetase, Peroxidase Unknown function: Putative actin-binding protein and putative splicing factor-like protein | [75] |
| Oryza sativa L. cv. Nipponbare and Zhonghua 8 | Osmotic Mannitol 400 mM, 48 h | Redox homeostasis: glutathione S-transferase (GST) Heat shock proteins: heat shock protein, dnaK-type molecular chaperone, endosperm luminal binding protein (BiP), Housekeeping: 26S proteasome regulatory subunit, Signal transduction: calreticulin precursor Lipid accumulation: lipid transfer protein, Glyoxalase–glyoxalase I Proteasome regulatory pathways: 20S proteasome α-subunit, proteasome-degradation system-related proteins, endoplasmic reticulum (ER)-related proteins Cell death-related protein: uroporphyrinogen decarboxylase | [89] |
| Oryza sativa L. | Osmotic 20% PEG, 8 days | Redox metabolism; Prx and putative thioredoxin peroxidase Photosynthesis-rbcS and rbcL Cytoskeleton stability: putative actin-binding protein, ABP Defense: putative chitinase Protein metabolism: ribonuclease Signal transduction: voltage-dependent anion selective channel protein and osmotin-like protein | [90] |
| Oryza sativa L. cv. Dongjin | Heat 42 °C, for 12 and 24 h | Heat shock proteins: HSP 70, dnak-type molecular chaperone, endosperm luminal binding protein (Bip), putative chaperonin 60 (Cpn 60) precusor Energy and metabolism: related protein-Transketolase, UDPglucose pyrophosphorylase, putative thiamine, pyruvate dehydrogenase complex (PDC) Redox homeostasis: GST, dehydro-ascorbate reductase (DHAR), thioredoxin h-type, SOD Regulatory proteins/ housekeeping enzymes: chloroplast elongation factors, cysteine proteinase, proteosome subunit alpha type1 and subunit of 20s proteosome, nucleoside diphosphate kinase 1 (NDPK1) | [91] |
| Oryza sativa L. ssp. japonica | Cold 15, 10 and 5 °C 24 h | Signal transduction: Elongation factor Metabolism synthesis: S-adenosylmethionine synthetase 2, VB12-independent methionine synthase Antioxidative: GDP-mannose 3′,5′-epimerase Protein metabolism: chaperonin, ATP-dependent Clp protease ATP-binding subunit Oxygen-evolving complex proteins: NADH-ubiquinone oxidoreductase, putative ferredoxin-NADP(H) oxidoreductase | [92] |
| Oryza sativa L. cv. Nipponbare | Cold 5 °C 48 h | Cellulose synthesis: UDP-glucose pyrophosphorylase Energy metabolism: adenylate kinase protein, RuBisCO LSU, vacuolar ATPase B subunit, H+ transporting ATP synthase, fructose-bisphosphate aldolase Protease: cysteine proteinase, 5-methyltetrahyropteroyltriglutamate-homocysteine S-methyl transferase, protein disulfide isomerase, Stress defense: Betaine aldehyde dehydrogenase (salt), Phenylalanine ammonia lyase (mechanical wounding), Beta-1,3-glucanases Signal transduction: Calreticulin, phosphoglycerate kinase, Elongation factor G Heat shock protein: HSP70 Housekeeping enzymes: nucleoside diphosphate kinase (NDPK) Antioxidant enzymes: superoxide dismutase (Cu/Zn), catalase, Unknown function: phosphoglucomutase, chitinase III-like protein, malate dehydrogenase | [93] |
| Oryza sativa L. cv. Dongjin | Cold 5 °C 12 h, 24 h, 36 h 10 °C 24 h and 72 h | Antioxidant enzymes: Ascorbate peroxidase, putative glutathione S-transferase, thioredoxin h-type (Thx h) and thioredoxin peroxidase Housekeeping enzymes: nucleoside diphosphate kinase 1 (NDPK1) Lipid-binding protein-fibrillin-like protein Protease: cysteine proteinase Regulatory: drought-inducible late embryogenesis abundant protein, RING zinc finger protein-like | [94] |
| Oryza sativa L. Chunyou 58 and Yongyou 6 | Nitrogen Shortage of N for 12 h, 3 days and 7 days | Photosynthetic metabolism: ribulose-1,5-bisphosphate carboxylase/oxygenase activase, type II tight-harvesting chlorophyll a/b-binding protein, carbonic anhydrases, rubisco large subunit, 23kDa polypeptide of photosystem II, dTDP-glucose 4–6-dehydratase-like protein and H protein subunit of glycine decarboxylase 3′-partial Stress responses/defenses: DegP2, harpin-binding proteins, heat shock-related proteins, glutathione S-transferase GSTF14, Fibrillin-like protein, Glyceraldehyde-3-phosphate dehydrogenase Membrane transporter: Putative chloroplast inner envelop protein, SecA protein | [95] |
| Oryza sativa L. cv. Dongjin | Chemical treatment 100 μM CdCl2 for 24 h | Antioxidant enzymes: l-ascorbate peroxidase 1, GR, glutathione S-transferases, NADH-ubiquinone oxidoreductase, hypothetical protein, peroxidase, putative ferredoxin-NADP(H) oxidoreductase Carbohydrate metabolism: Bisphosphoglycerate-independent phosphoglycerate mutase, glyceraldehyde-3-phosphate dehydrogenase, Alpha-1,4-glucan-protein synthase, endo-1,3-betaglucanase Amino acids and photosynthesis metabolism: glutamine synthetase, Photosystem II oxygen-evolving complex protein 2, ribulose bisphosphate carboxylase/oxygenase activase Protein metabolism: Putative ubiquitin isopeptidase T, 26S proteasome, Chloroplast translational elongation factor Tu, elongation factor P, Putative chaperonin 60 beta, vacuolar proton-ATPase, guanine nucleotide-binding protein subunit beta-like protein, ricin B-related lectin domain-containing protein | [96] |
| Oryza sativa L. cv. Nipponbare | Ozone 0.2 ppm, 24 h | Cellular processing and signaling: Ion transporters, MAPK, Ca2+-dependent protein kinase (CPKs), Ca2+-binding proteins, receptor kinases Photosynthesis: ATP-dependent Clp protease, chloroplast cell division protease ftsH homologous, HSP 90, Rubisco Defense: chloroplast L-APX, glutathione peroxidase, putative basic secretory protein Antioxidant: glutathione S-transferase, glutathione peroxidase, glutathione reductase, catalase, monodehydroacorbate reductase | [68] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, N.L.; Rahmat, Z.; Lam, S.S. A Review of the “Omics” Approach to Biomarkers of Oxidative Stress in Oryza sativa. Int. J. Mol. Sci. 2013, 14, 7515-7541. https://doi.org/10.3390/ijms14047515
Ma NL, Rahmat Z, Lam SS. A Review of the “Omics” Approach to Biomarkers of Oxidative Stress in Oryza sativa. International Journal of Molecular Sciences. 2013; 14(4):7515-7541. https://doi.org/10.3390/ijms14047515
Chicago/Turabian StyleMa, Nyuk Ling, Zaidah Rahmat, and Su Shiung Lam. 2013. "A Review of the “Omics” Approach to Biomarkers of Oxidative Stress in Oryza sativa" International Journal of Molecular Sciences 14, no. 4: 7515-7541. https://doi.org/10.3390/ijms14047515
APA StyleMa, N. L., Rahmat, Z., & Lam, S. S. (2013). A Review of the “Omics” Approach to Biomarkers of Oxidative Stress in Oryza sativa. International Journal of Molecular Sciences, 14(4), 7515-7541. https://doi.org/10.3390/ijms14047515