Frataxin Deficiency Leads to Reduced Expression and Impaired Translocation of NF-E2-Related Factor (Nrf2) in Cultured Motor Neurons

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
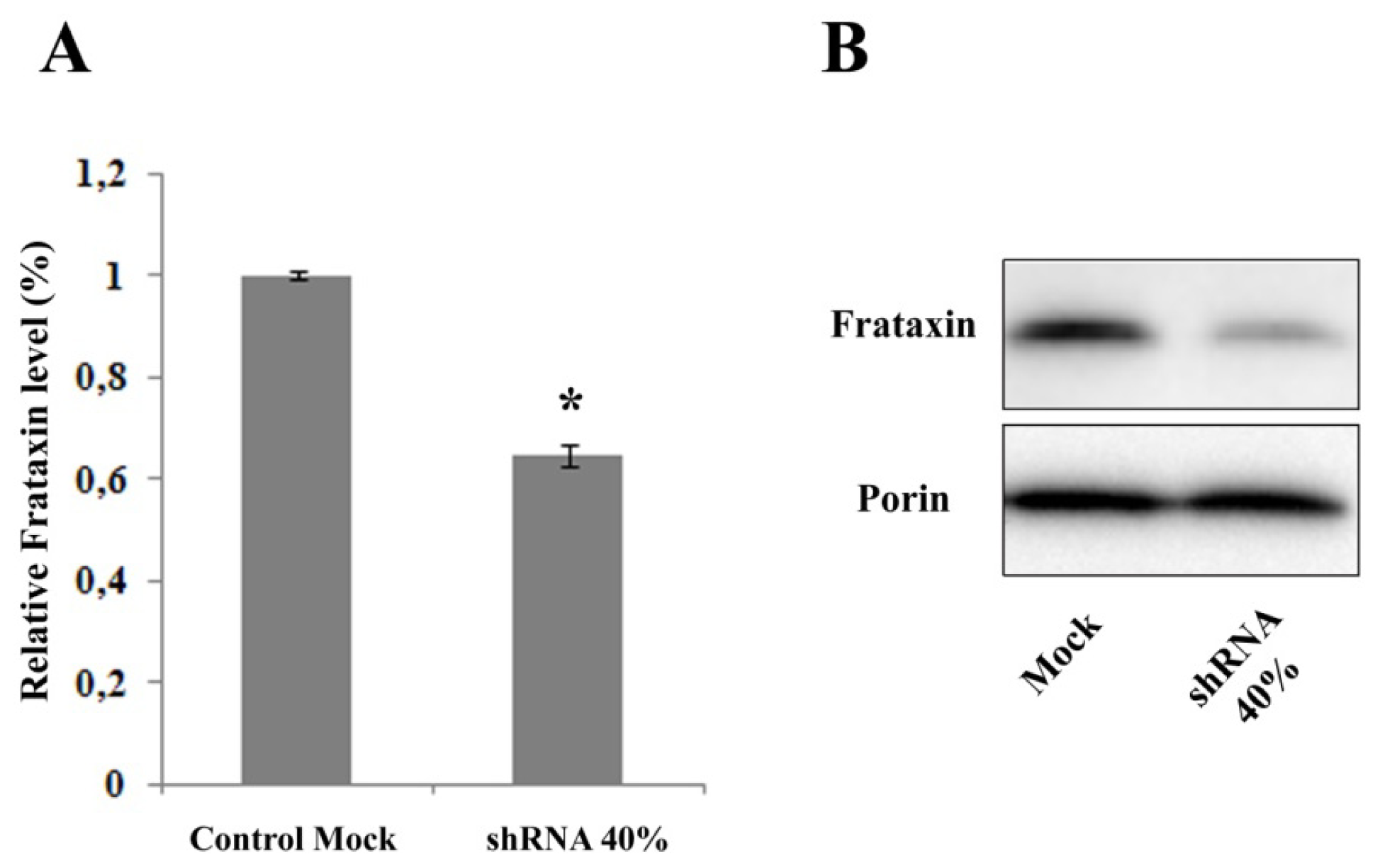
2.1. Frataxin Silencing in NSC34 Motor Neurons
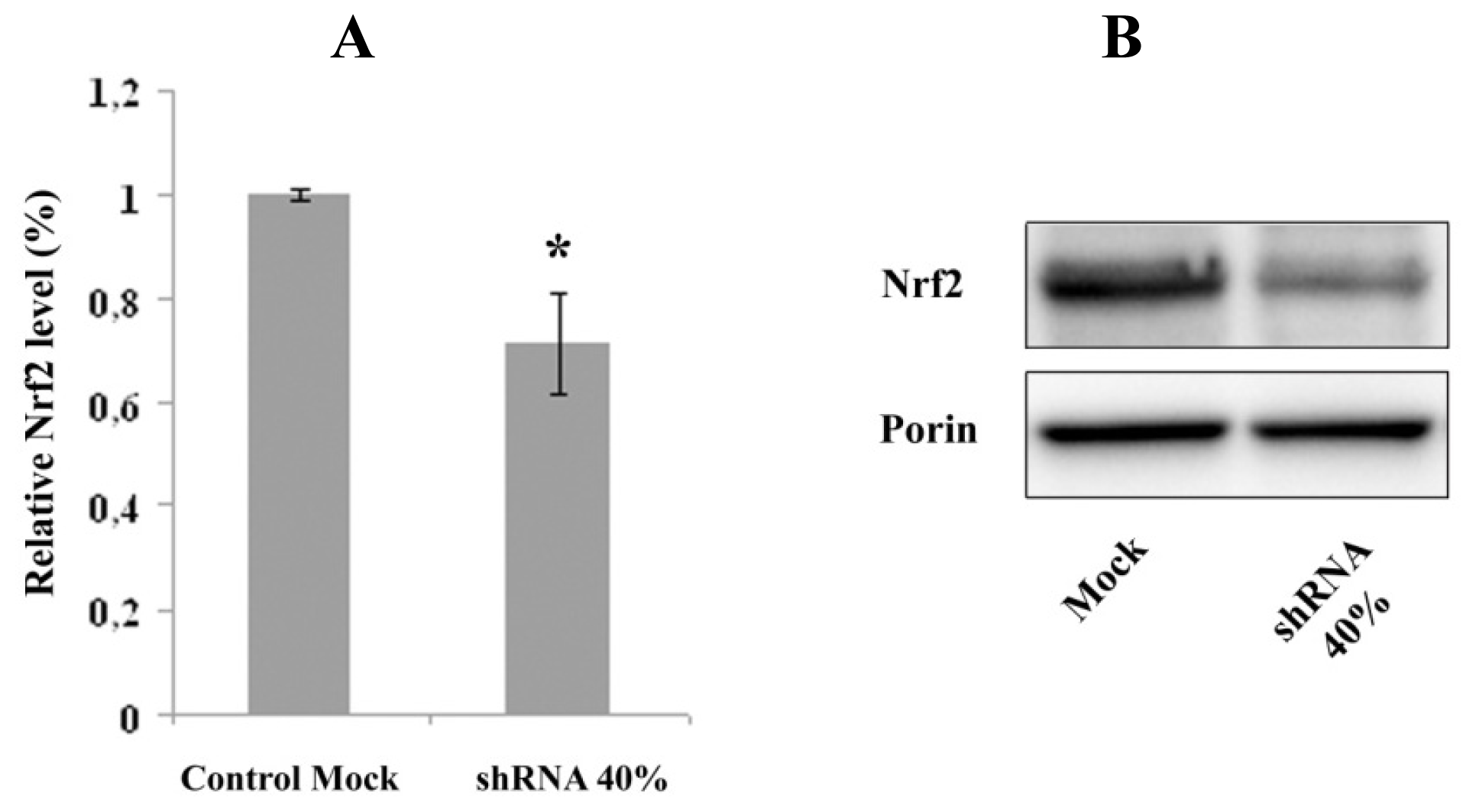
2.2. Nrf2 Expression Is Decreased in Frataxin-Silenced Neurons
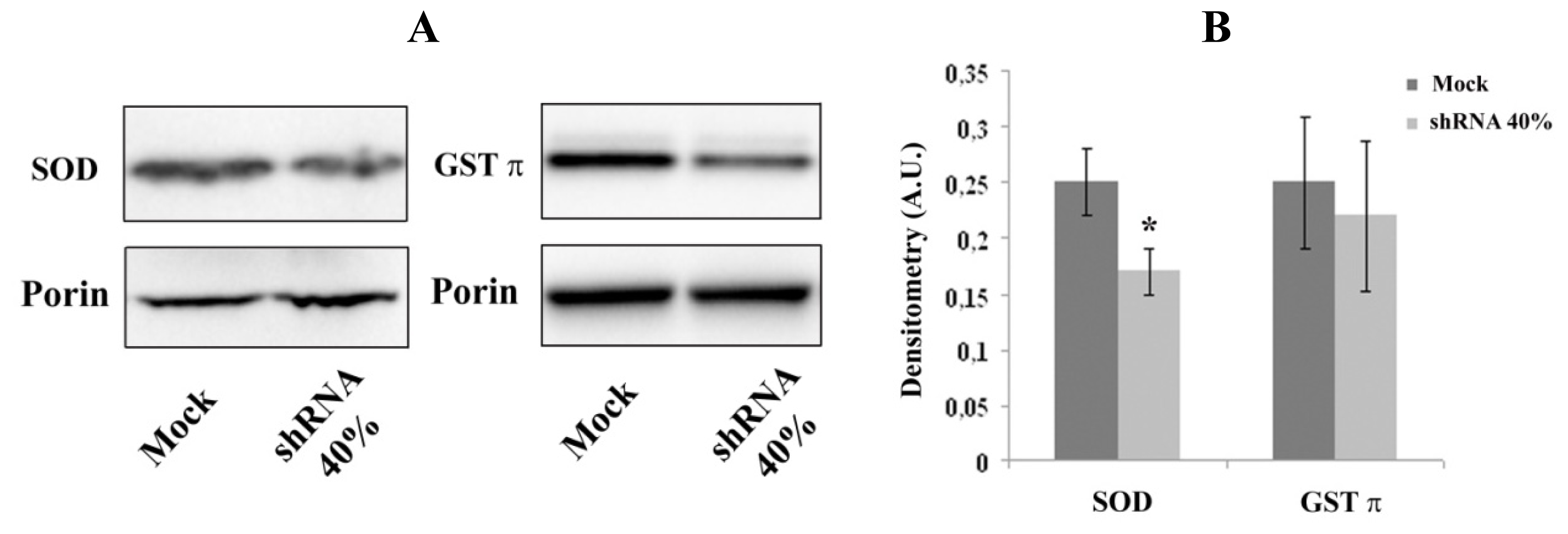
2.3. Decreased Expression of Nrf2-Targeted Genes in Frataxin-Silenced Neurons
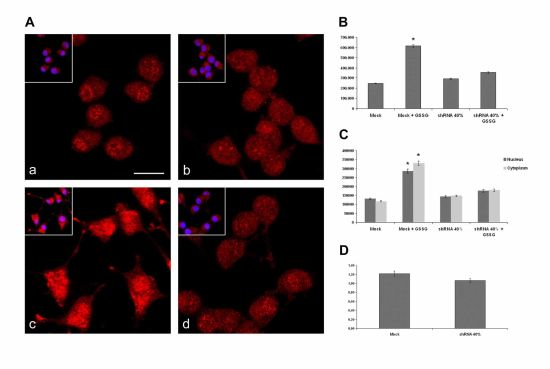
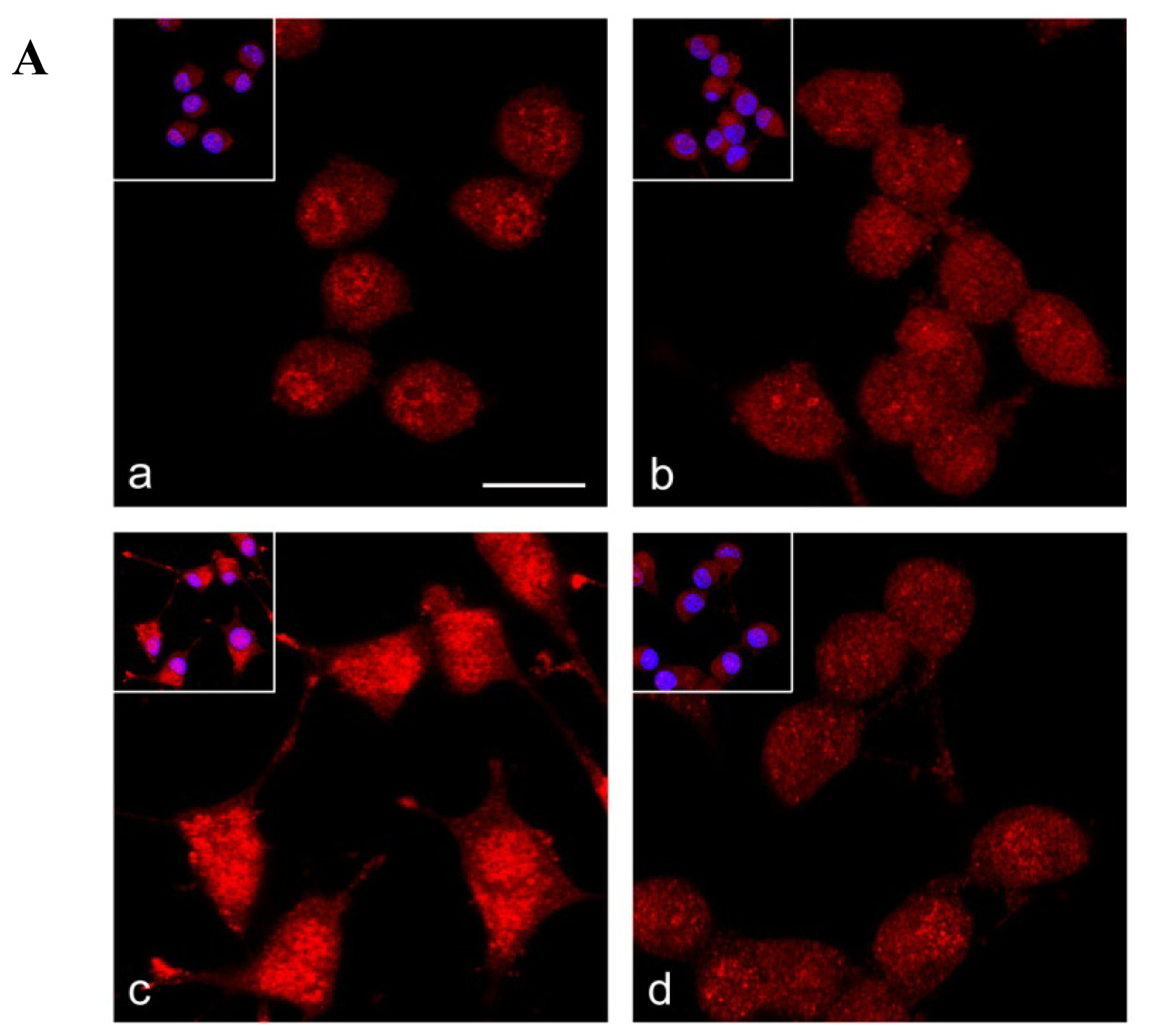
2.4. Nrf2 Distribution in Frataxin-Silenced Neurons
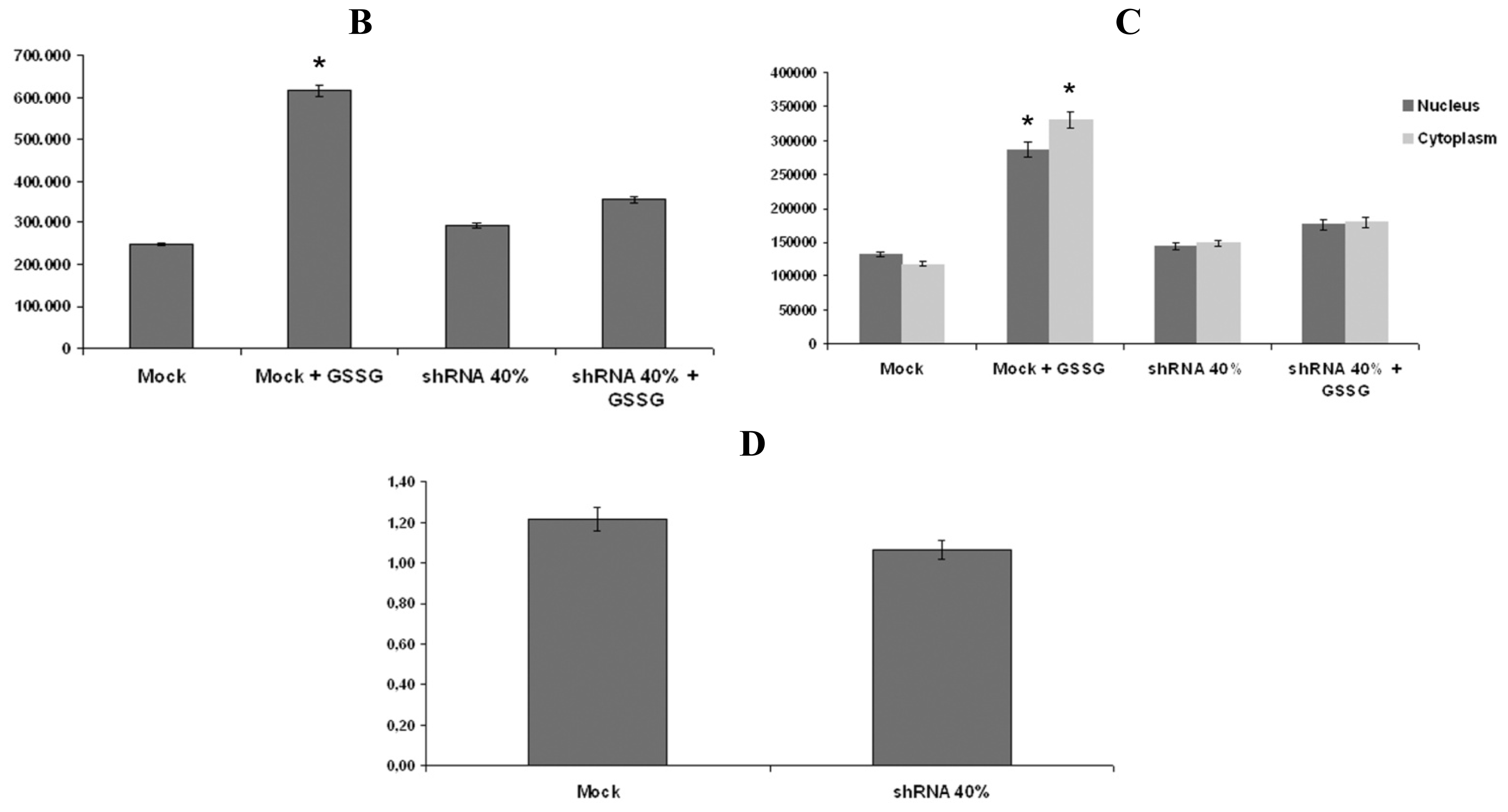
2.5. Nrf2 Fails to Translocate to Frataxin Silenced Neurons Nuclei in Response to Oxidative Stress
2.6. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Stable shRNA Cell Lines Generation
3.3. Real-Time Quantitative RT-PCR
3.4. Western Blot Analysis of Frataxin and Nrf2 Protein Levels
3.5. Western Blot Analysis of Nrf2 and Downstream Target Proteins
3.6. Immunocytochemistry
3.7. Imaging Analysis
3.8. Statistical Analysis
4. Conclusions
Abbreviations
| FRDA | Friedreich’s Ataxia |
| Nrf2 | NF-E2-related factor |
| ARE | antioxidant response element |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
Conflict of Interest
References
- Oppenheimer, D.; Esiri, M. Disease of the Basal Ganglia, Cerebellum and Motor Neurons. In Greenfield’s Neuropathology, 5th ed; Adams, J.H., Corsellis, J., Duchen, L.W., Eds.; Arnold: London, UK, 1992; pp. 988–1054. [Google Scholar]
- Fogel, B.L.; Perlman, S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 2007, 6, 245–257. [Google Scholar]
- Alper, G.; Narayanan, V. Friedreich’s ataxia. Pediatr. Neurol 2003, 28, 335–341. [Google Scholar]
- Sakamato, N.; Chastain, P.D.; Parniewski, P.; Ohshima, K.; Pandolfo, M.; Griffith, J.D.; Wells, R.D. Sticky DNA: Self-association properties of long GAA TTC repeats in RR Y triplex structures from Friedreich’s ataxia. Mol. Cell 1999, 3, 465–475. [Google Scholar]
- Santos, R.; Lefevre, S.; Sliwa, D.; Seguin, A.; Camadro, J.M.; Lesuisse, E. Friedreich ataxia: Molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid. Redox Signal 2010, 13, 651–690. [Google Scholar]
- Pandolfo, M. Friedreich ataxia: New pathways. J. Child. Neurol 2012, 27, 1204–1211. [Google Scholar]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci 2005, 233, 145–162. [Google Scholar]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet 2000, 9, 275–282. [Google Scholar]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat. Genet 1997, 17, 215–217. [Google Scholar]
- Lynch, D.R.; Lech, G.; Farmer, J.M.; Balcer, L.J.; Bank, W.; Chance, B.; Wilson, R.B. Near infrared muscle spectroscopy in patients with Friedreich’s ataxia. Muscle Nerve 2002, 25, 664–673. [Google Scholar]
- Vorgerd, M.; Schols, L.; Hardt, C.; Ristow, M.; Epplen, J.T.; Zange, J. Mitochondrial impairment of human muscle in Friedreich ataxia in vivo. Neuromuscul. Disord 2000, 10, 430–435. [Google Scholar]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar]
- Schulz, J.B.; Dehmer, T.; Schols, L.; Mender, H.; Hardt, C.; Vorgerd, M.; Burk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 719–721. [Google Scholar]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Invest 2001, 31, 1007–1011. [Google Scholar]
- Tozzi, G.; Nuccetelli, M.; Lo Bello, M.; Bernardini, S.; Bellincampi, L.; Ballerini, S.; Gaeta, L.M.; Casali, C.; Pastore, A.; Federici, G.; et al. Antioxidant enzymes in blood of patients with Friedreich’s ataxia. Arch. Dis. Child 2002, 86, 376–379. [Google Scholar]
- Martelli, A.; Napierala, M.; Puccio, H. Understanding the genetic and molecular pathogenesis of Friedreich’s ataxia through animal and cellular models. Dis. Model Mech 2012, 5, 165–176. [Google Scholar]
- Bulteau, A.L.; Dancis, A.; Gareil, M.; Montagne, J.J.; Camadro, J.M.; Lesuisse, E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med 2007, 42, 1561–1570. [Google Scholar]
- Auchere, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet 2008, 17, 2790–2802. [Google Scholar]
- Lefevre, S.; Brossas, C.; Auchere, F.; Boggetto, N.; Camadro, J.M.; Santos, R. Apn1 AP-endonuclease is essential for the repair of oxidatively damaged DNA bases in yeast frataxin-deficient cells. Hum. Mol. Genet 2012, 21, 4060–4072. [Google Scholar]
- Vazquez-Manrique, R.P.; Gonzalez-Cabo, P.; Ros, S.; Aziz, H.; Baylis, H.A.; Palau, F. Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. FASEB J 2006, 20, 172–174. [Google Scholar]
- Runko, A.P.; Griswold, A.J.; Min, K.T. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett 2008, 582, 715–719. [Google Scholar]
- Anderson, P.R.; Kirby, K.; Hilliker, A.J.; Phillips, J.P. RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum. Mol. Genet 2005, 14, 3397–3405. [Google Scholar]
- Anderson, P.R.; Kirby, K.; Orr, W.C.; Hilliker, A.J.; Phillips, J.P. Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich’s ataxia. Proc. Natl. Acad. Sci. USA 2008, 105, 611–616. [Google Scholar]
- Ristow, M.; Mulder, H.; Pomplun, D.; Schulz, T.J.; Muller-Schmehl, K.; Krause, A.; Fex, M.; Puccio, H.; Muller, J.; Isken, F.; et al. Frataxin deficiency in pancreatic islets causes diabetes due to loss of beta cell mass. J. Clin. Invest 2003, 112, 527–534. [Google Scholar]
- Thierbach, R.; Schulz, T.J.; Isken, F.; Voigt, A.; Mietzner, B.; Drewes, G.; von Kleist-Retzow, J.C.; Wiesner, R.J.; Magnuson, M.A.; Puccio, H.; et al. Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum. Mol. Genet 2005, 14, 3857–3864. [Google Scholar]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.M.; Li, J.; Johnson, J.A. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal 2009, 11, 497–508. [Google Scholar]
- Prestera, T.; Talalay, P.; Alam, J.; Ahm, Y.I.; Lee, P.J.; Choi, A.M. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: Regulation by upstream antioxidant-responsive elements (ARE). Mol. Med 1995, 1, 827–837. [Google Scholar]
- Favreau, L.V.; Pickett, C.B. The rat quinone reductase antioxidant response element: Identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and nonhepatoma cell lines. J. Biol. Chem 1995, 270, 24468–24474. [Google Scholar]
- Wang, B.; Williamson, G. Detection of a nuclear protein that binds specifically to the antioxidant responsive element (ARE) of the human NAD(P)H:quinone oxidoreductase gene. Biochim. Biophys. Acta 1994, 1219, 645–652. [Google Scholar]
- Rushmore, T.H.; Pickett, C.B. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene: Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J. Biol. Chem 1990, 265, 14648–14653. [Google Scholar]
- Galloway, D.C.; Blake, D.G.; Shepherd, A.G.; McLellan, L.I. Regulation of human γ-glutamylcysteine synthetase: Coordinate induction of the catalytic and regulatory subunits in HepG2 cells. Biochem. J 1997, 328, 99–104. [Google Scholar]
- Galloway, D.C.; McLellan, L.I. Inducible expression of the γ-glutamylcysteine synthetase light subunit by t-butylhydroquinone in HepG2 cells is not dependent on an antioxidant-responsive element. Biochem. J 1998, 336, 535–539. [Google Scholar]
- Mulcahy, R.T.; Gipp, J.J. Identification of a putative antioxidant response element in the 5-flanking region of the human γ-glutamylcysteine synthetase heavy subunit gene. Biochem. Biophys. Res. Commun 1995, 209, 227–233. [Google Scholar]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi-Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, in press. [Google Scholar]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One 2009, 4, e4253. [Google Scholar]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol 2013, 85, 705–717. [Google Scholar]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci 2008, 1147, 61–69. [Google Scholar]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol 1998, 150, 60–68. [Google Scholar]
- Yoo, M.S.; Chun, H.S.; Son, J.J.; DeGiorgio, L.A.; Kim, D.J.; Peng, C.; Son, J.H. Oxidative stress-regulated genes in nigral dopaminergic neuronal cells: Correlation with the known pathology in Parkinson’s disease. Brain Res. Mol. Brain Res 2003, 110, 76–84. [Google Scholar]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci 2008, 28, 13574–13581. [Google Scholar]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar]
- Haugen, A.C.; Di Prospero, N.A.; Parker, J.S.; Fannin, R.D.; Chou, J.; Meyer, J.N.; Halweg, C.; Collins, J.B.; Durr, A.; Fischbeck, K.; et al. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: Cellular model of pathology. PLoS Genet 2010, 6, e1000812. [Google Scholar]
- Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Nuclear import and export signals in control of Nrf2. J. Biol. Chem 2005, 280, 29158–29168. [Google Scholar]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem 2003, 278, 42588–42595. [Google Scholar]
- Townsend, D.M. S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Mol. Interv 2007, 7, 313–324. [Google Scholar]
- Carletti, B.; Passarelli, C.; Sparaco, M.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. Effect of protein glutathionylation on neuronal cytoskeleton: A potential link to neurodegeneration. Neuroscience 2011, 192, 285–294. [Google Scholar]





© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D'Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin Deficiency Leads to Reduced Expression and Impaired Translocation of NF-E2-Related Factor (Nrf2) in Cultured Motor Neurons. Int. J. Mol. Sci. 2013, 14, 7853-7865. https://doi.org/10.3390/ijms14047853
D'Oria V, Petrini S, Travaglini L, Priori C, Piermarini E, Petrillo S, Carletti B, Bertini E, Piemonte F. Frataxin Deficiency Leads to Reduced Expression and Impaired Translocation of NF-E2-Related Factor (Nrf2) in Cultured Motor Neurons. International Journal of Molecular Sciences. 2013; 14(4):7853-7865. https://doi.org/10.3390/ijms14047853
Chicago/Turabian StyleD'Oria, Valentina, Stefania Petrini, Lorena Travaglini, Chiara Priori, Emanuela Piermarini, Sara Petrillo, Barbara Carletti, Enrico Bertini, and Fiorella Piemonte. 2013. "Frataxin Deficiency Leads to Reduced Expression and Impaired Translocation of NF-E2-Related Factor (Nrf2) in Cultured Motor Neurons" International Journal of Molecular Sciences 14, no. 4: 7853-7865. https://doi.org/10.3390/ijms14047853
APA StyleD'Oria, V., Petrini, S., Travaglini, L., Priori, C., Piermarini, E., Petrillo, S., Carletti, B., Bertini, E., & Piemonte, F. (2013). Frataxin Deficiency Leads to Reduced Expression and Impaired Translocation of NF-E2-Related Factor (Nrf2) in Cultured Motor Neurons. International Journal of Molecular Sciences, 14(4), 7853-7865. https://doi.org/10.3390/ijms14047853