Inhibition of CCL2 Signaling in Combination with Docetaxel Treatment Has Profound Inhibitory Effects on Prostate Cancer Growth in Bone

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
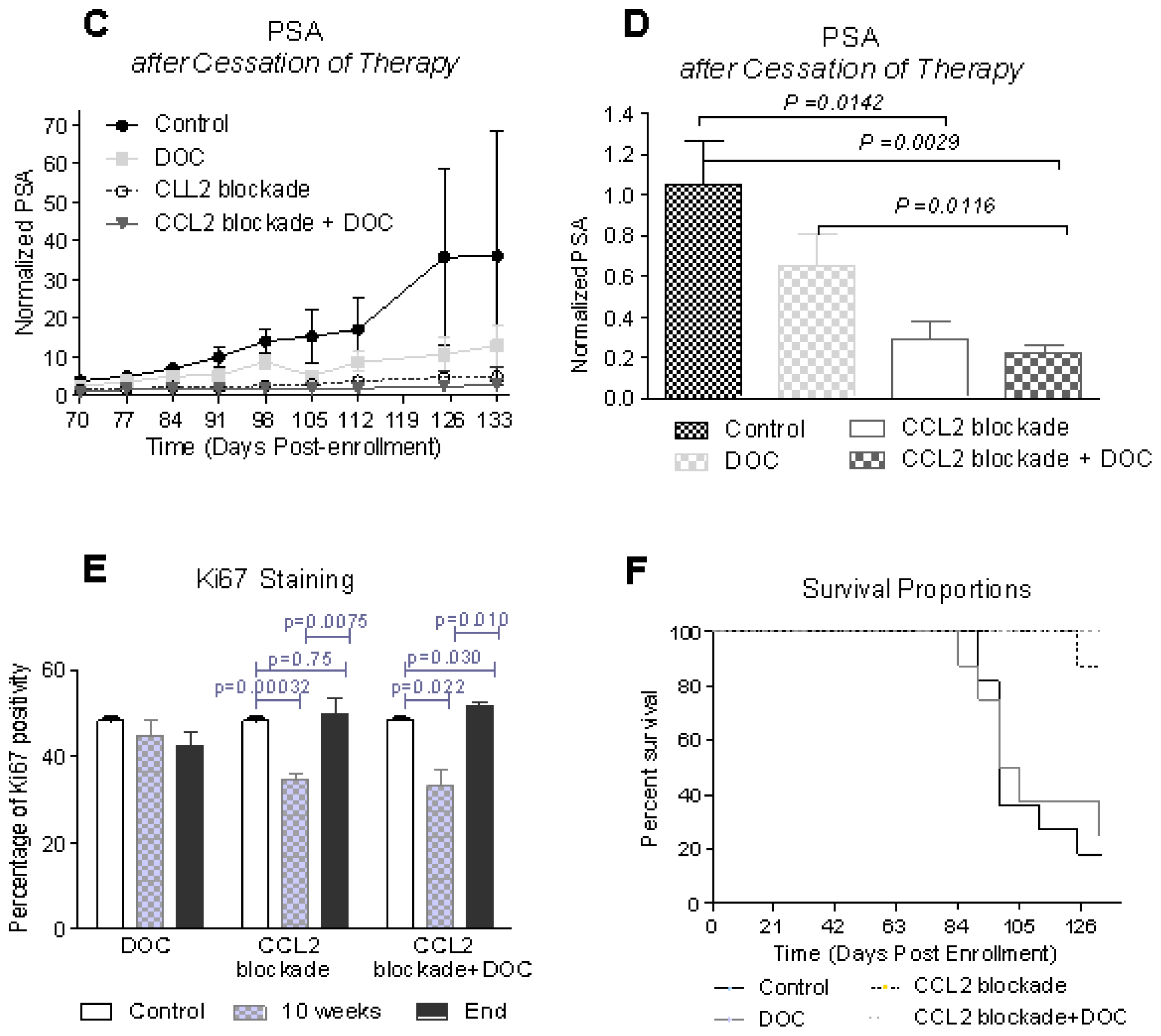
2.1. CCL2 Blockade Inhibits Prostate Tumor Progression in Bone
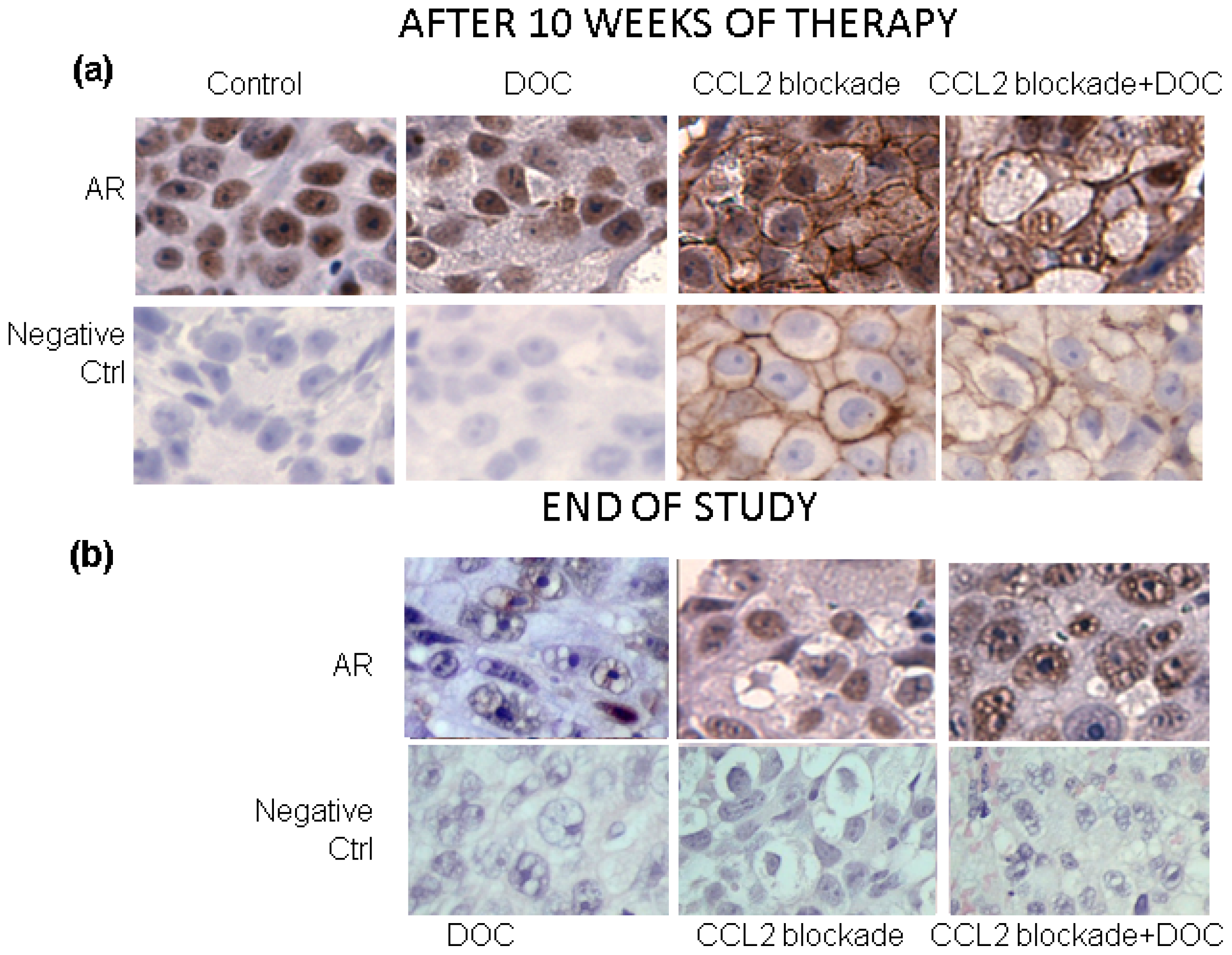
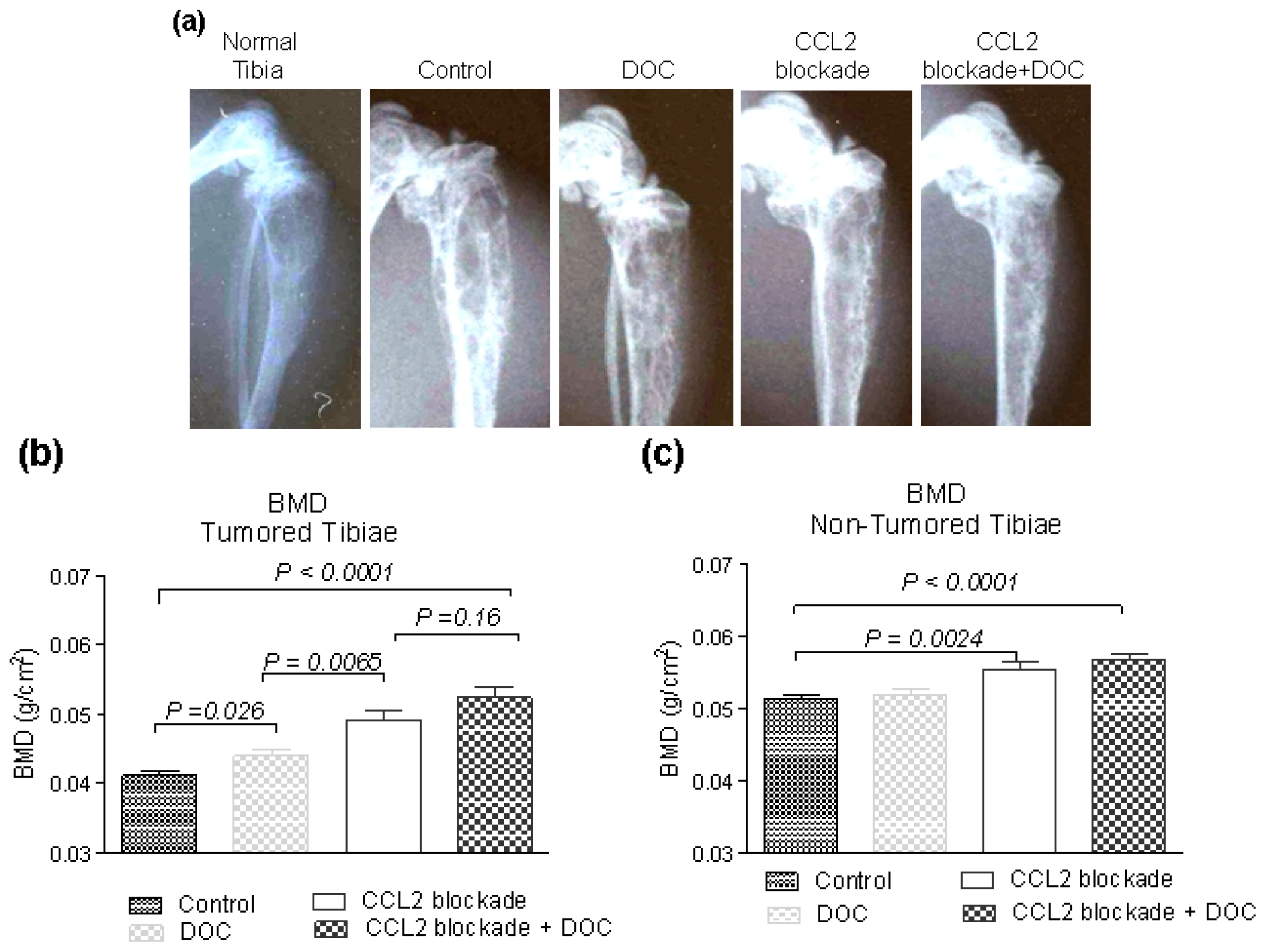
2.2. Effects of CCL2 Blockade on Tumor-Bearing Bone
2.3. Effects of CCL2 Blockade on Normal Bone
2.4. Effects of CCL2 Blockade on Bodyweight
2.5. Implications
3. Experimental Section
3.1. Animal Experiments
3.2. Immunohistochemistry (IHC)
3.3. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin 2009, 59, 225–249. [Google Scholar]
- Petrylak, D. Therapeutic options in androgen-independent prostate cancer: Building on docetaxel. BJU Int 2005, 96, 41–46. [Google Scholar]
- Armstrong, A.J.; Garrett-Mayer, E.S.; Yang, Y.C.; de Wit, R.; Tannock, I.F.; Eisenberger, M. A contemporary prognostic nomogram for men with hormone-refractory metastatic prostate cancer: A TAX327 study analysis. Clin. Cancer Res 2007, 13, 6396–6403. [Google Scholar]
- Berthold, D.R.; Pond, G.R.; Soban, F.; de Wit, R.; Eisenberger, M.; Tannock, I.F. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: Updated survival in the TAX 327 study. J. Clin. Oncol 2008, 26, 242–245. [Google Scholar]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med 2004, 351, 1502–1512. [Google Scholar]
- Lu, Y.; Chen, Q.; Corey, E.; Xie, W.; Fan, J.; Mizokami, A.; Zhang, J. Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin. Exp. Metastasis 2009, 26, 161–169. [Google Scholar]
- Shirotake, S.; Miyajima, A.; Kosaka, T.; Tanaka, N.; Kikuchi, E.; Mikami, S.; Okada, Y.; Oya, M. Regulation of monocyte chemoattractant protein-1 through angiotensin II type 1 receptor in prostate cancer. Am. J. Pathol 2012, 180, 1008–1016. [Google Scholar]
- Sun, T.; Mary, L.G.; Oh, W.K.; Freedman, M.L.; Pomerantz, M.; Pienta, K.J.; Kantoff, P.W. Inherited variants in the chemokine CCL2 gene and prostate cancer aggressiveness in a Caucasian cohort. Clin. Cancer Res 2011, 17, 1546–1552. [Google Scholar]
- Shi, C.L.; Yu, C.H.; Zhang, Y.; Zhao, D.; Chang, X.H.; Wang, W.H. Monocyte chemoattractant protein-1 modulates invasion and apoptosis of PC-3M prostate cancer cells via regulating expression of VEGF, MMP9 and caspase-3. Asian Pac. J. Cancer Prev 2011, 12, 555–559. [Google Scholar]
- Mizutani, K.; Sud, S.; McGregor, N.A.; Martinovski, G.; Rice, B.T.; Craig, M.J.; Varsos, Z.S.; Roca, H.; Pienta, K.J. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia 2009, 11, 1235–1242. [Google Scholar]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res 2007, 67, 9417–9424. [Google Scholar]
- Craig, M.; Ying, C.; Loberg, R.D. Co-inoculation of prostate cancer cells with U937 enhances tumor growth and angiogenesis in vivo. J. Cell Biochem 2008, 103, 1–8. [Google Scholar]
- Li, X.; Loberg, R.; Liao, J.; Ying, C.; Snyder, L.A.; Pienta, K.J.; McCauley, L.K. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res 2009, 69, 1685–1692. [Google Scholar]
- Fulton, A.M. The chemokine receptors CXCR4 and CXCR3 in cancer. Curr. Oncol. Rep 2009, 11, 125–131. [Google Scholar]
- Leber, M.F.; Efferth, T. Molecular principles of cancer invasion and metastasis (review). Int. J. Oncol 2009, 34, 881–895. [Google Scholar]
- Rubin, J.B. Chemokine signaling in cancer: One hump or two? Semin. Cancer Biol 2009, 19, 116–122. [Google Scholar]
- Wu, X.; Lee, V.C.; Chevalier, E.; Hwang, S.T. Chemokine receptors as targets for cancer therapy. Curr. Pharm. Des 2009, 15, 742–757. [Google Scholar]
- Waugh, D.J.; Wilson, C.; Seaton, A.; Maxwell, P.J. Multi-faceted roles for CXC-chemokines in prostate cancer progression. Front. Biosci 2008, 13, 4595–4604. [Google Scholar]
- Vindrieux, D.; Escobar, P.; Lazennec, G. Emerging roles of chemokines in prostate cancer. Endocr. Relat Cancer 2009, 16, 663–673. [Google Scholar]
- Lu, Y.; Cai, Z.; Galson, D.L.; Xiao, G.; Liu, Y.; George, D.E.; Melhem, M.F.; Yao, Z.; Zhang, J. Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate 2006, 66, 1311–1318. [Google Scholar]
- Lu, Y.; Cai, Z.; Xiao, G.; Keller, E.T.; Mizokami, A.; Yao, Z.; Roodman, G.D.; Zhang, J. Monocyte chemotactic protein-1 mediates prostate cancer-induced bone resorption. Cancer Res 2007, 67, 3646–3653. [Google Scholar]
- Loberg, R.D.; Ying, C.; Craig, M.; Yan, L.; Snyder, L.A.; Pienta, K.J. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia 2007, 9, 556–562. [Google Scholar]
- Izhak, L.; Wildbaum, G.; Zohar, Y.; Anunu, R.; Klapper, L.; Elkeles, A.; Seagal, J.; Yefenof, E.; yalon-Soffer, M.; Karin, N. A novel recombinant fusion protein encoding a 20-amino acid residue of the third extracellular (E3) domain of CCR2 neutralizes the biological activity of CCL2. J. Immunol 2009, 183, 732–739. [Google Scholar]
- Zollo, M.; di Dato, V.; Spano, D.; de Martino, D.; Liguori, L.; Marino, N.; Vastolo, V.; Navas, L.; Garrone, B.; Mangano, G.; et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin. Exp. Metastasis 2012, 29, 585–601. [Google Scholar]
- Zhang, J.; Lu, Y.; Pienta, K.J. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst 2010, 102, 522–528. [Google Scholar]
- Zhang, J.; Patel, L.; Pienta, K.J. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev 2010, 21, 41–48. [Google Scholar]
- Zhang, J.; Patel, L.; Pienta, K.J. Targeting chemokine (C-C motif) ligand 2 (CCL2) as an example of translation of cancer molecular biology to the clinic. Prog. Mol. Biol. Transl. Sci 2010, 95, 31–53. [Google Scholar]
- Binder, N.B.; Niederreiter, B.; Hoffmann, O.; Stange, R.; Pap, T.; Stulnig, T.M.; Mack, M.; Erben, R.G.; Smolen, J.S.; Redlich, K. Estrogen-dependent and C-C chemokine receptor-2-dependent pathways determine osteoclast behavior in osteoporosis. Nat. Med 2009, 15, 417–424. [Google Scholar]
- Qian, D.Z.; Rademacher, B.L.; Pittsenbarger, J.; Huang, C.Y.; Myrthue, A.; Higano, C.S.; Garzotto, M.; Nelson, P.S.; Beer, T.M. CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity. Prostate 2010, 70, 433–442. [Google Scholar]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest. New Drugs 2012, 31, 760–768. [Google Scholar]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.W.K. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res 1994, 54, 2577–2581. [Google Scholar]
- Pfitzenmaier, J.; Quinn, J.E.; Odman, A.M.; Zhang, J.; Keller, E.T.; Vessella, R.L.; Corey, E. Characterization of C4-2 prostate cancer bone metastases and their response to castration. J. Bone Miner. Res 2003, 18, 1882–1888. [Google Scholar]
- Corey, E.; Quinn, J.E.; Bladou, F.; Brown, L.G.; Roudier, M.P.; Brown, J.M.; Buhler, K.R.; Vessella, R.L. Establishment and characterization of osseous prostate cancer models: Intra-tibial injection of human prostate cancer cells. Prostate 2002, 52, 20–33. [Google Scholar]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar]
- Sarafi, M.N.; Garcia-Zepeda, E.A.; MacLean, J.A.; Charo, I.F.; Luster, A.D. Murine monocyte chemoattractant protein (MCP)-5: A novel CC chemokine that is a structural and functional homologue of human MCP-1. J. Exp. Med 1997, 185, 99–109. [Google Scholar]
- Corey, E.; Quinn, J.E.; Buhler, K.R.; Nelson, P.S.; Macoska, J.A.; True, L.D.; Vessella, R.L. LuCaP 35: A new model of prostate cancer progression to androgen independence. Prostate 2003, 55, 239–246. [Google Scholar]





© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kirk, P.S.; Koreckij, T.; Nguyen, H.M.; Brown, L.G.; Snyder, L.A.; Vessella, R.L.; Corey, E. Inhibition of CCL2 Signaling in Combination with Docetaxel Treatment Has Profound Inhibitory Effects on Prostate Cancer Growth in Bone. Int. J. Mol. Sci. 2013, 14, 10483-10496. https://doi.org/10.3390/ijms140510483
Kirk PS, Koreckij T, Nguyen HM, Brown LG, Snyder LA, Vessella RL, Corey E. Inhibition of CCL2 Signaling in Combination with Docetaxel Treatment Has Profound Inhibitory Effects on Prostate Cancer Growth in Bone. International Journal of Molecular Sciences. 2013; 14(5):10483-10496. https://doi.org/10.3390/ijms140510483
Chicago/Turabian StyleKirk, Peter S., Theodore Koreckij, Holly M. Nguyen, Lisha G. Brown, Linda A. Snyder, Robert L. Vessella, and Eva Corey. 2013. "Inhibition of CCL2 Signaling in Combination with Docetaxel Treatment Has Profound Inhibitory Effects on Prostate Cancer Growth in Bone" International Journal of Molecular Sciences 14, no. 5: 10483-10496. https://doi.org/10.3390/ijms140510483
APA StyleKirk, P. S., Koreckij, T., Nguyen, H. M., Brown, L. G., Snyder, L. A., Vessella, R. L., & Corey, E. (2013). Inhibition of CCL2 Signaling in Combination with Docetaxel Treatment Has Profound Inhibitory Effects on Prostate Cancer Growth in Bone. International Journal of Molecular Sciences, 14(5), 10483-10496. https://doi.org/10.3390/ijms140510483