Turn-Directed α-β Conformational Transition of α-syn12 Peptide at Different pH Revealed by Unbiased Molecular Dynamics Simulations


Abstract
:1. Introduction
2. Results and Discussion
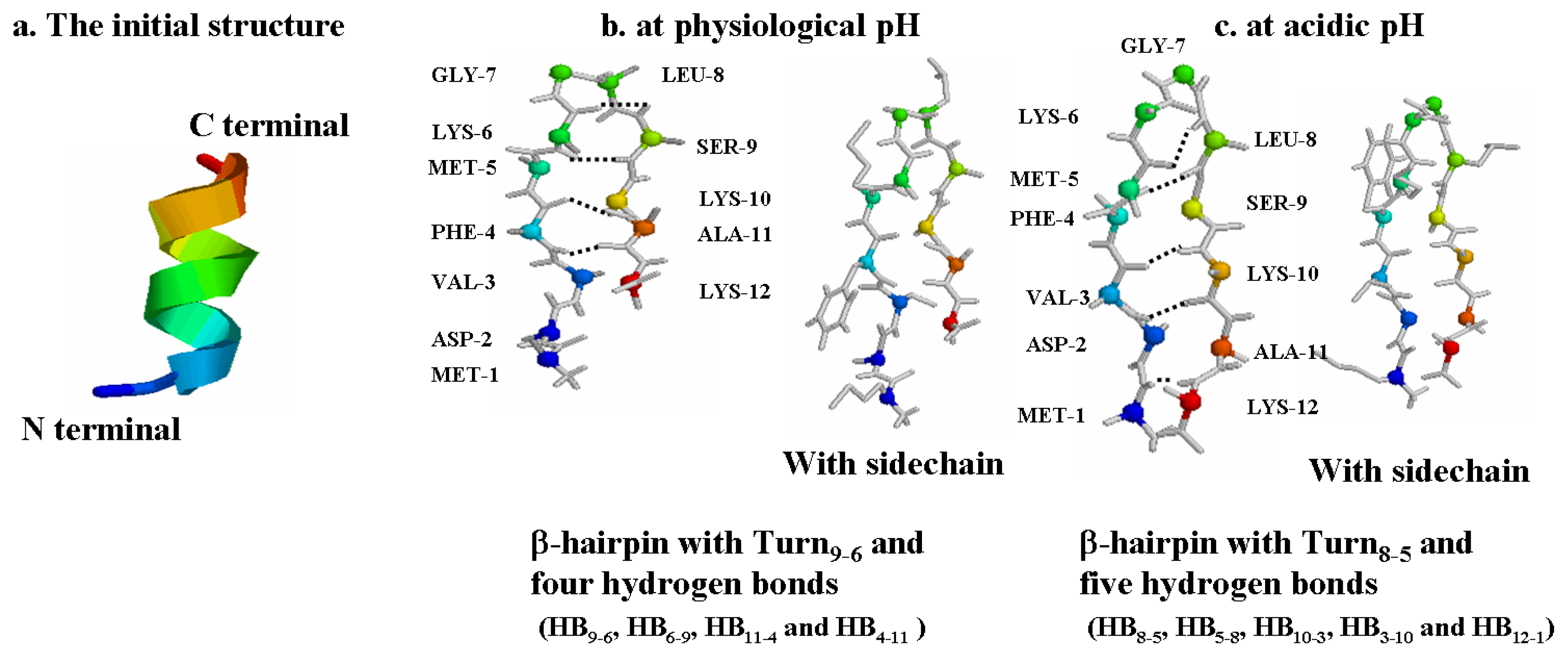
2.1. The Most Clustered Structures
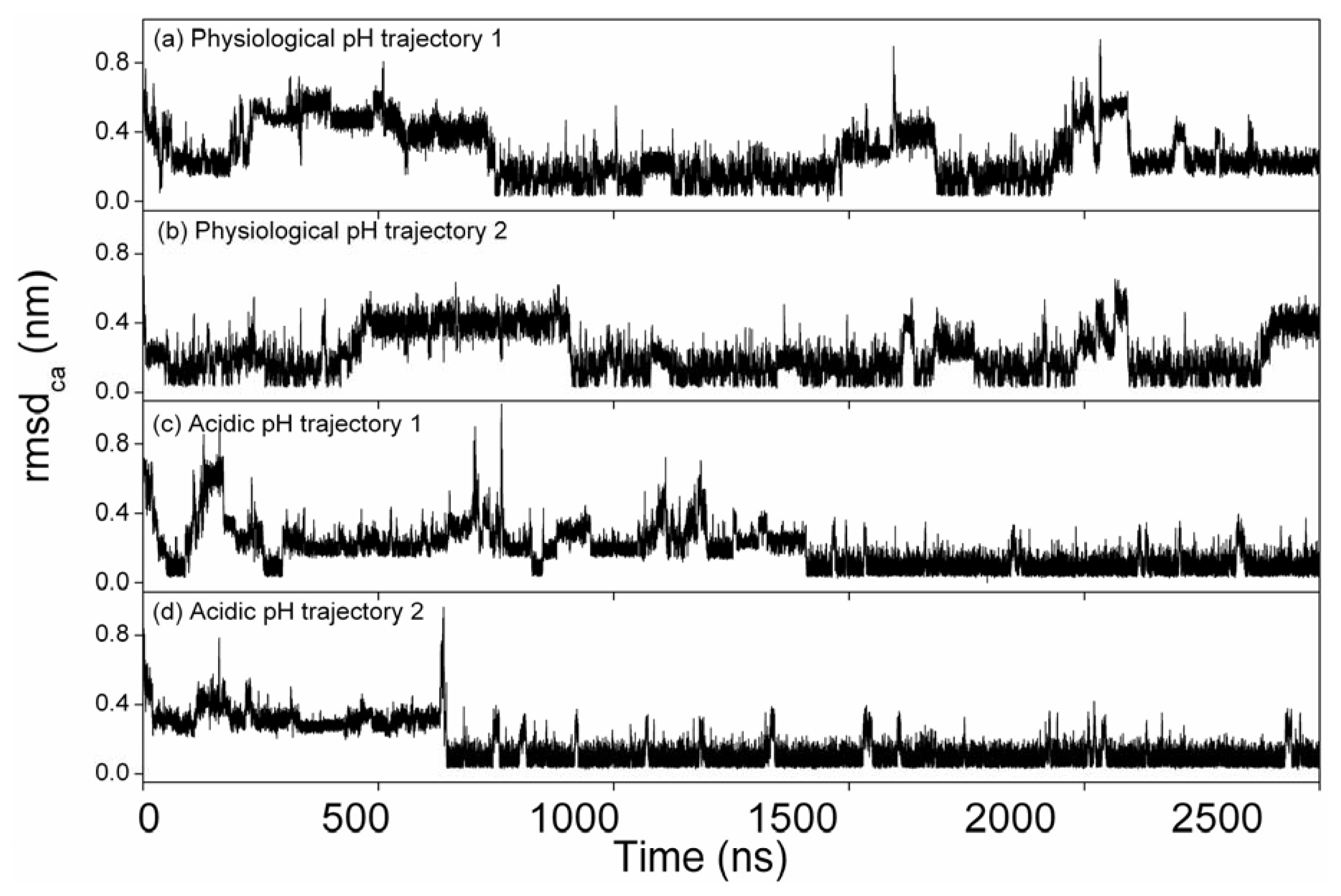
2.2. Root Mean Square Deviations of Cα Atoms (RMSDCα)
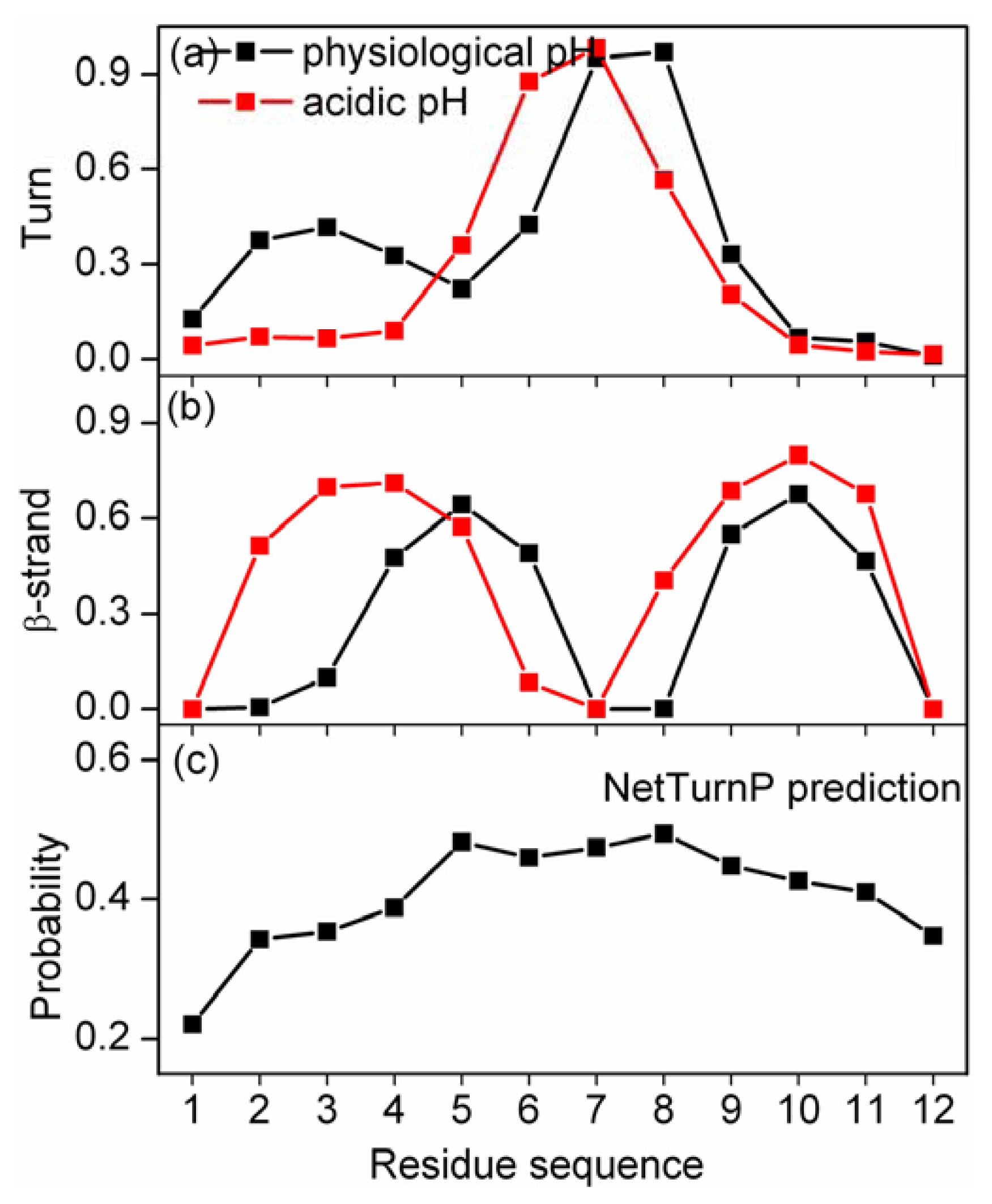
2.3. Residue-Specific Secondary Structure Propensity
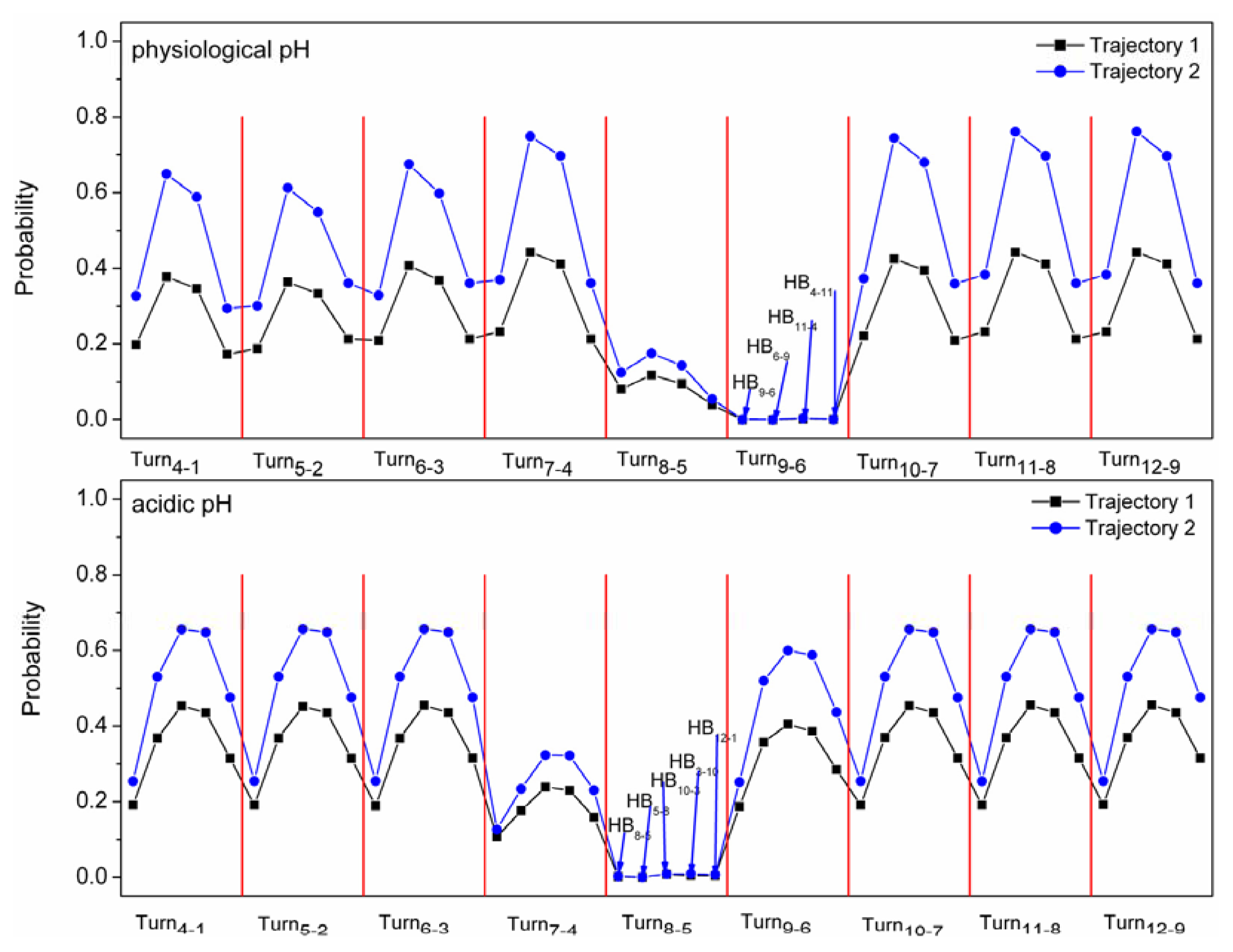
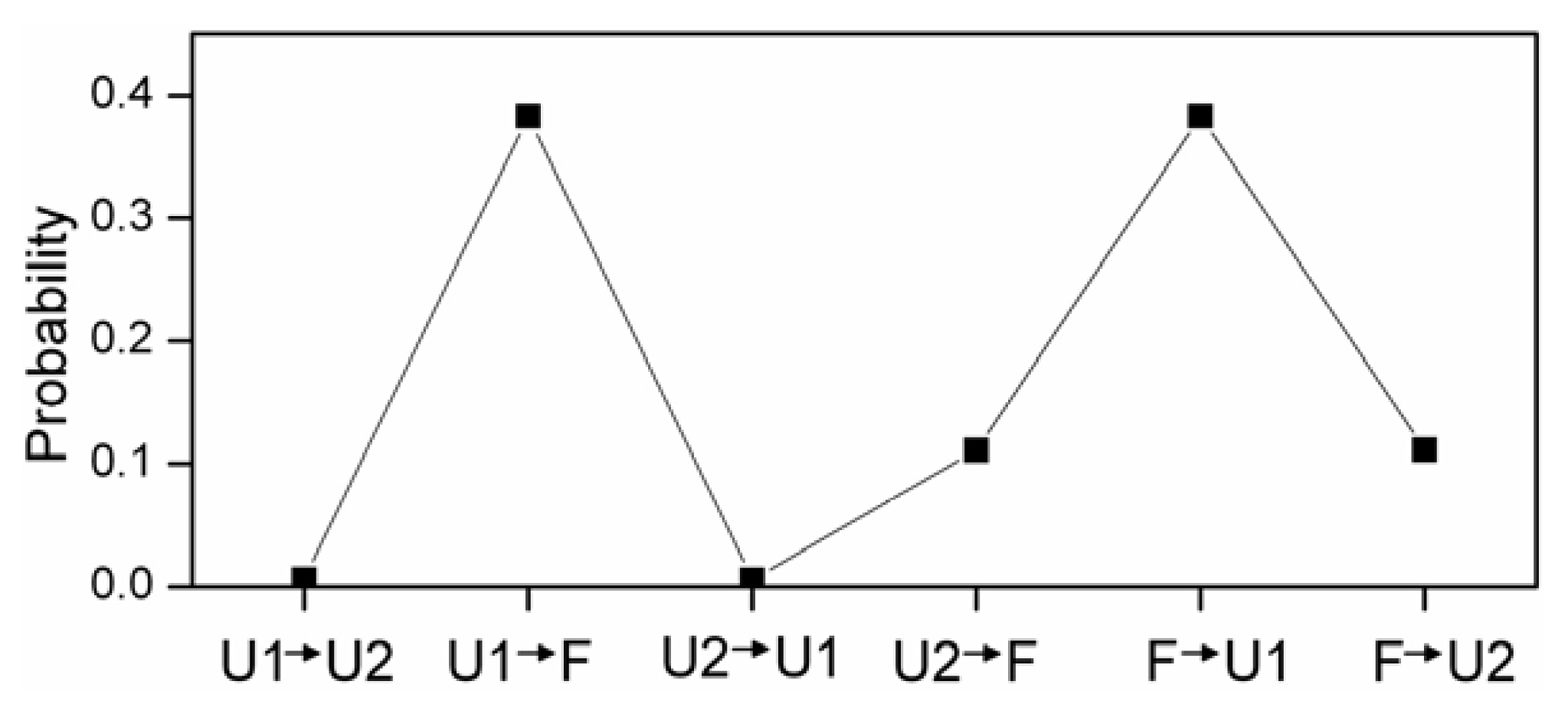
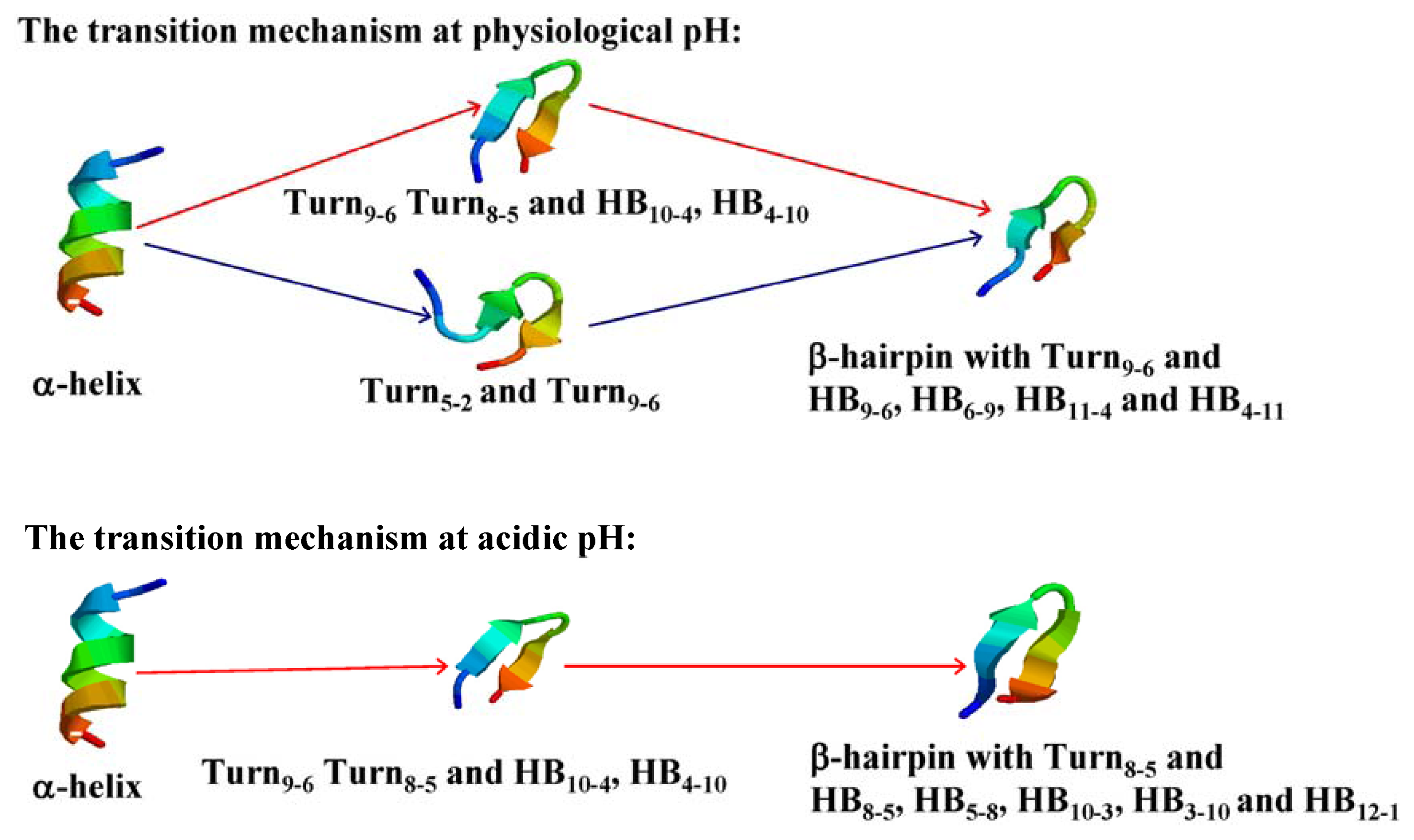
2.4. The Relationship between Turns and Hydrogen Bonds
2.5. Free Energy Surface and the Transition Path
3. Methods
4. Conclusions
Supplementary Information
ijms-14-10896-s001.pdfAcknowledgments
Conflict of Interest
References
- Bisaglia, M.; Mammi, S.; Bubacco, L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J 2009, 23, 329–340. [Google Scholar]
- Yoon, J.; Jang, S.; Lee, K.; Shin, S. Simulation studies on the stabilities of aggregates formed by fibril-forming segments of alpha-Synuclein. J. Biomol. Struct. Dyn 2009, 27, 259–270. [Google Scholar]
- Yoshiki, Y.; Masami, M.; Hiroaki, S.; Takashi, N.; Shinya, H.; Shin-ichi, H.; Koichi, K.; Masato, H. Characterization of inhibitor-bound α-synuclein dimer: Role of α-synuclein N-terminal region in dimerization and inhibitor binding. J. Mol. Biol 2010, 395, 445–456. [Google Scholar]
- Xie, Y.Y.; Zhou, C.J.; Zhou, Z.R.; Hong, J.; Che, M.X.; Fu, Q.S.; Song, A.X.; Lin, D.H.; Hu, H.Y. Interaction with synphilin-1 promotes inclusion formation of {alpha}-synuclein: Mechanistic insights and pathological implication. FASEB J 2009, 24, 196–205. [Google Scholar]
- Cao, Z.; Liu, L.; Wu, P.; Wang, J. Structural and thermodynamics characters of isolated α-syn12 peptide: Long-time temperature replica-exchange molecular dynamics in aqueous solution. Acta Biochim. Biophys. Sin 2011, 43, 172–180. [Google Scholar]
- Cao, Z.; Liu, L.; Wang, J. Effects of pH and temperature on the structural and thermodynamic character of alpha-syn12 peptide in aqueous solution. J. Biomol. Struct. Dyn 2010, 28, 343–353. [Google Scholar]
- Cao, Z.; Liu, L.; Zhao, L.; Li, H.; Wang, J. Comparison of the structural characteristics of Cu(2+)-bound and unbound alpha-syn12 peptide obtained in simulations using different force fields. J. Mol. Model 2013, 19, 1237–1250. [Google Scholar]
- Dyer, R.B.; Maness, S.J.; Peterson, E.S.; Franzen, S.; Fesinmeyer, R.M.; Andersen, N.H. The mechanism of beta-hairpin formation. Biochemistry 2004, 43, 11560–11566. [Google Scholar]
- Evans, D.A.; Wales, D.J. Folding of the GB1 hairpin peptide from discrete path sampling. J. Chem. Phys 2004, 121, 1080–1090. [Google Scholar]
- Wu, X.; Brooks, B.R. Beta-hairpin folding mechanism of a nine-residue peptide revealed from molecular dynamics simulations in explicit water. Biophys. J 2004, 86, 1946–1958. [Google Scholar]
- Munoz, V.; Ghirlando, R.; Blanco, F.J.; Jas, G.S.; Hofrichter, J.; Eaton, W.A. Folding and aggregation kinetics of a beta-hairpin. Biochemistry 2006, 45, 7023–7035. [Google Scholar]
- Nguyen, P.H.; Stock, G.; Mittag, E.; Hu, C.K.; Li, M.S. Free energy landscape and folding mechanism of a beta-hairpin in explicit water: A replica exchange molecular dynamics study. Proteins 2005, 61, 795–808. [Google Scholar]
- Pande, V.S.; Rokhsar, D.S. Molecular dynamics simulations of unfolding and refolding of a beta-hairpin fragment of protein G. Proc. Natl. Acad. Sci. USA 1999, 96, 9062–9067. [Google Scholar]
- Munoz, V.; Thompson, P.A.; Hofrichter, J.; Eaton, W.A. Folding dynamics and mechanism of beta-hairpin formation. Nature 1997, 390, 196–199. [Google Scholar]
- Thukral, L.; Smith, J.C.; Daidone, I. Common folding mechanism of a beta-hairpin peptide via non-native turn formation revealed by unbiased molecular dynamics simulations. J. Am. Chem. Soc 2009, 131, 18147–18152. [Google Scholar]
- Thukral, L.; Daidone, I.; Smith, J.C. Structured pathway across the transition state for peptide folding revealed by molecular dynamics simulations. PLoS Comput. Biol 2011, 7, e1002137. [Google Scholar]
- Yoda, T.; Sugita, Y.; Okamoto, Y. Cooperative folding mechanism of a beta-hairpin peptide studied by a multicanonical replica-exchange molecular dynamics simulation. Proteins 2007, 66, 846–859. [Google Scholar]
- Daidone, I.; Simona, F.; Roccatano, D.; Broglia, R.A.; Tiana, G.; Colombo, G.; di Nola, A. Beta-hairpin conformation of fibrillogenic peptides: Structure and alpha-beta transition mechanism revealed by molecular dynamics simulations. Proteins 2004, 57, 198–204. [Google Scholar]
- Daidone, I.; Amadei, A.; di Nola, A. Thermodynamic and kinetic characterization of a beta-hairpin peptide in solution: An extended phase space sampling by molecular dynamics simulations in explicit water. Proteins 2005, 59, 510–518. [Google Scholar]
- Daidone, I.; Ulmschneider, M.B.; di Nola, A.; Amadei, A.; Smith, J.C. Dehydration-driven solvent exposure of hydrophobic surfaces as a driving force in peptide folding. Proc. Natl. Acad. Sci. USA 2007, 104, 15230–15235. [Google Scholar]
- Chiang, Y.W.; Otoshima, Y.; Watanabe, Y.; Inanami, O.; Shimoyama, Y. Dynamics and local ordering of spin-labeled prion protein: An ESR simulation study of a highly PH-sensitive site. J. Biomol. Struct. Dyn 2008, 26, 355–366. [Google Scholar]
- Levy, Y.; Hanan, E.; Solomon, B.; Becker, O.M. Helix-coil transition of PrP106–126: Molecular dynamic study. Proteins 2001, 45, 382–396. [Google Scholar]
- Klimov, D.K.; Thirumalai, D. Dissecting the assembly of Abeta16-22 amyloid peptides into antiparallel beta sheets. Structure 2003, 11, 295–307. [Google Scholar]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem 2001, 276, 10737–10744. [Google Scholar]
- Wu, K.P.; Weinstock, D.S.; Narayanan, C.; Levy, R.M.; Baum, J. Structural reorganization of alpha-synuclein at low pH observed by NMR and REMD simulations. J. Biol. Chem 2009, 391, 784–796. [Google Scholar]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res 2004, 32, W500–W502. [Google Scholar]
- Eichenberger, A.P.; Allison, J.R.; Dolenc, J.; Geerke, D.P.; Horta, B.A.C.; Meier, K.; Oostenbrink, C.; Schmid, N.; Steiner, D.; Wang, D.; et al. GROMOS++ software for the analysis of biomolecular simulation trajectories. J. Chem. Theory Comput 2011, 7, 3379–3390. [Google Scholar]
- Petersen, B.; Lundegaard, C.; Petersen, T.N. NetTurnP—Neural network prediction of beta-turns by use of evolutionary information and predicted protein sequence features. PLoS One 2010, 5, e15079. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem 2005, 26, 1701–1718. [Google Scholar]
- Van Gunsteren, W.F.; Billeter, S.R.; Eising, A.A.; Hunenberger, P.H.; Krüger, P.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. The GROMOS96 Manual and User Guide; Biomolecular Simulation: Zürich, Switzerland, 1996. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. Int. Forces 1981, 331–342. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8592. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126, 014101. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput 2008, 4, 116–122. [Google Scholar]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Turn9-6 | HB9-6 | HB6-9 | HB11-4 | HB4-11 | ||
|---|---|---|---|---|---|---|
| Trajectory 1 at physiological pH | 0.81 | 0.23 | 0.44 | 0.41 | 0.21 | |
| Trajectory 2 at physiological pH | 0.98 | 0.38 | 0.76 | 0.70 | 0.36 | |
| Turn8-5 | HB8-5 | HB5-8 | HB10-3 | HB3-10 | HB12-1 | |
| Trajectory 1 at physiological pH | 0.86 | 0.19 | 0.37 | 0.46 | 0.44 | 0.31 |
| Trajectory 2 at physiological pH | 0.66 | 0.18 | 0.36 | 0.45 | 0.44 | 0.48 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, L.; Cao, Z. Turn-Directed α-β Conformational Transition of α-syn12 Peptide at Different pH Revealed by Unbiased Molecular Dynamics Simulations. Int. J. Mol. Sci. 2013, 14, 10896-10907. https://doi.org/10.3390/ijms140610896
Liu L, Cao Z. Turn-Directed α-β Conformational Transition of α-syn12 Peptide at Different pH Revealed by Unbiased Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2013; 14(6):10896-10907. https://doi.org/10.3390/ijms140610896
Chicago/Turabian StyleLiu, Lei, and Zanxia Cao. 2013. "Turn-Directed α-β Conformational Transition of α-syn12 Peptide at Different pH Revealed by Unbiased Molecular Dynamics Simulations" International Journal of Molecular Sciences 14, no. 6: 10896-10907. https://doi.org/10.3390/ijms140610896
APA StyleLiu, L., & Cao, Z. (2013). Turn-Directed α-β Conformational Transition of α-syn12 Peptide at Different pH Revealed by Unbiased Molecular Dynamics Simulations. International Journal of Molecular Sciences, 14(6), 10896-10907. https://doi.org/10.3390/ijms140610896