UV Radiation and the Skin

Abstract
:1. The Skin
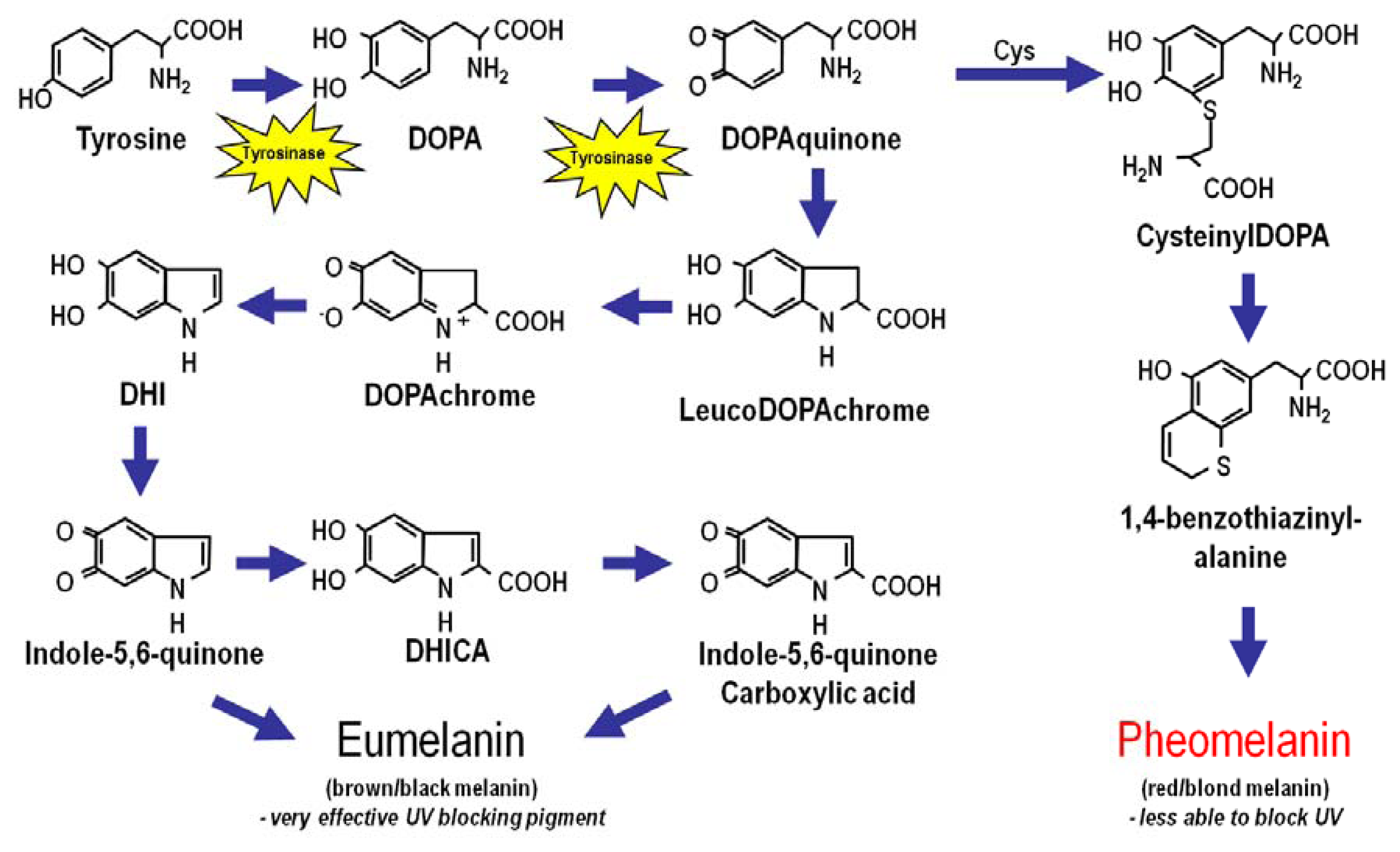
2. Melanin
3. Skin Pigmentation
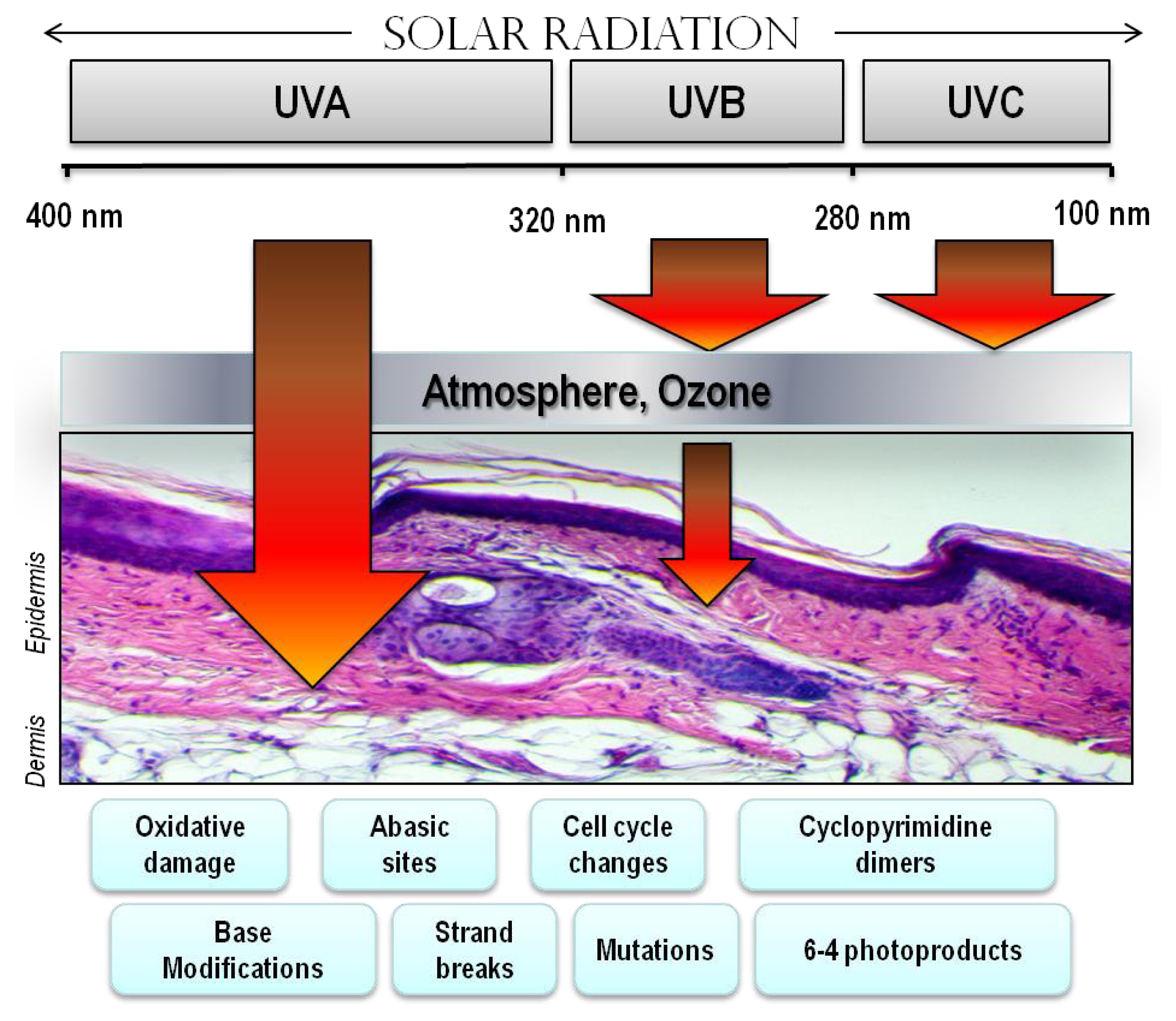
4. Ultraviolet Radiation (UV)
5. Indoor Tanning
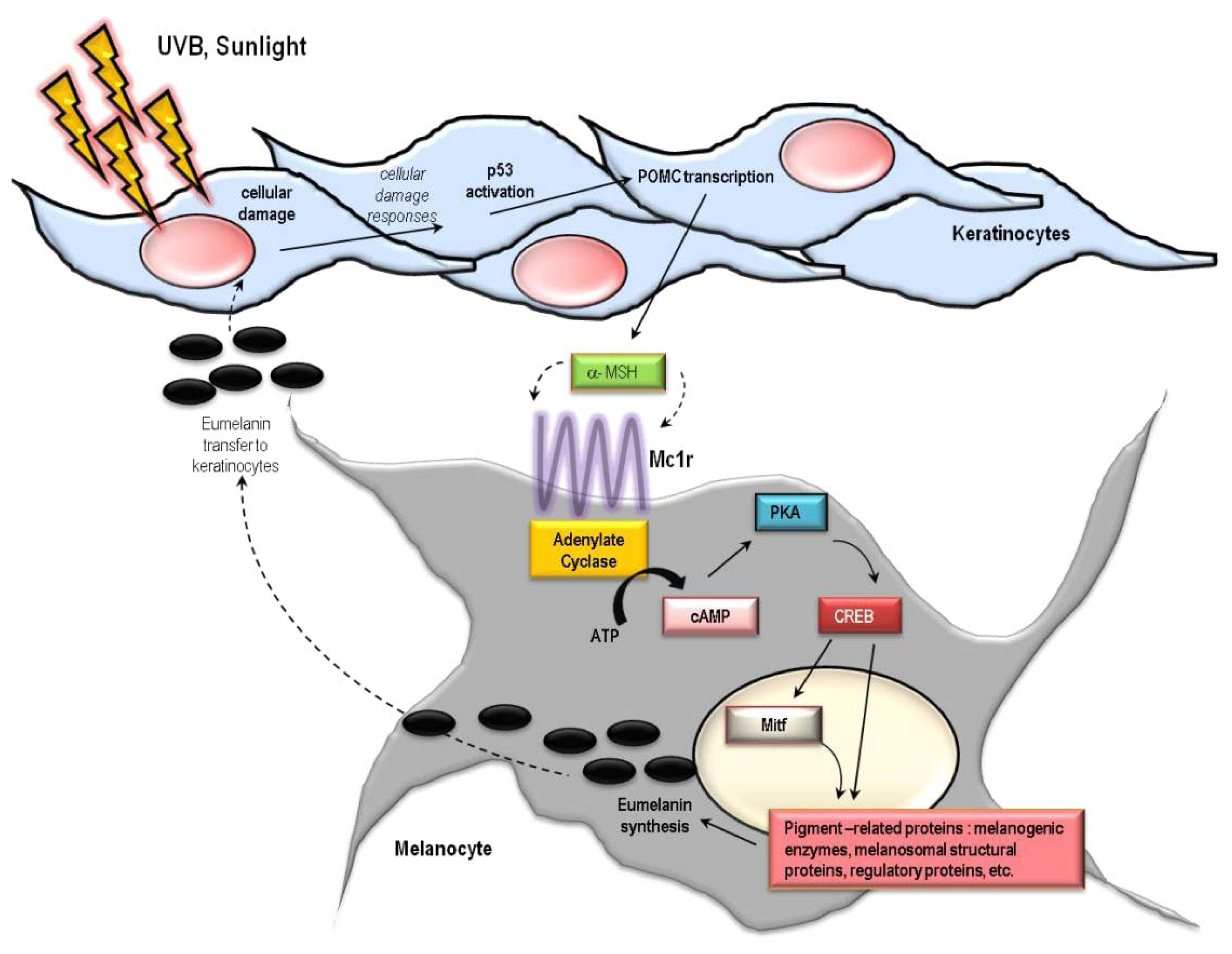
6. Cutaneous Responses to UV
7. Oxidative Injury
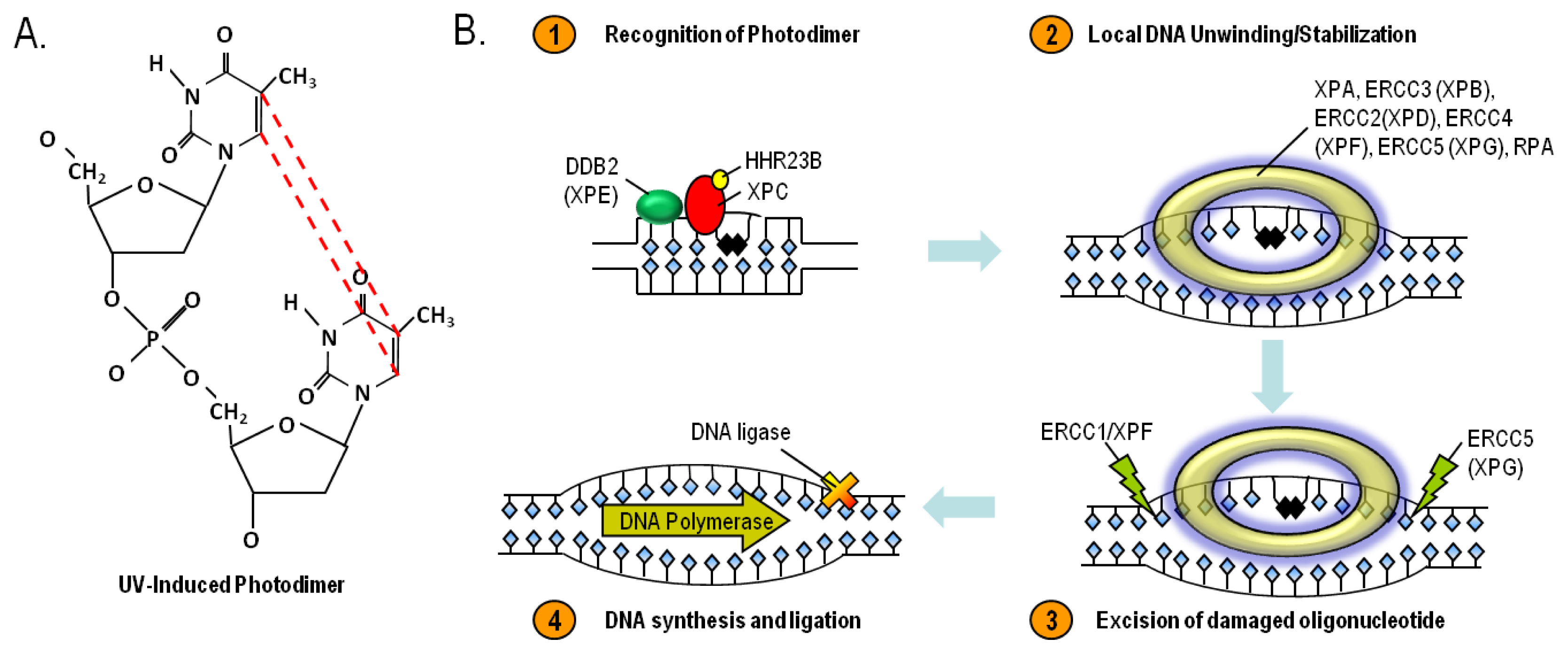
8. Nucleotide Excision Repair and Xeroderma Pigmentosum
9. Skin Cancer
10. The Melanocortin 1 Receptor (MC1R)
11. Conclusions
Acknowledgments
Conflicts of Interest
References
- Elwood, J.M.; Jopson, J. Melanoma and sun exposure: An overview of published studies. Int. J. Cancer 1997, 73, 198–203. [Google Scholar]
- Lowe, N.J. An overview of ultraviolet radiation, sunscreens, and photo-induced dermatoses. Dermatol. Clin 2006, 24, 9–17. [Google Scholar]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the skin. Endocr. Rev 2000, 21, 457–487. [Google Scholar]
- Slominski, A.; Wortsman, J.; Luger, T.; Paus, R.; Solomon, S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev 2000, 80, 979–1020. [Google Scholar]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar]
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell. Biol. 2012, 212, v-115. [Google Scholar]
- Fuchs, E.; Raghavan, S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet 2002, 3, 199–209. [Google Scholar]
- Madison, K.C. Barrier function of the skin: “la raison d'etre” of the epidermis. J. Invest. Dermatol 2003, 121, 231–241. [Google Scholar]
- Proksch, E.; Brandner, J.M.; Jensen, J.M. The skin: An indispensable barrier. Exp. Dermatol 2008, 17, 1063–1072. [Google Scholar]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev 2004, 84, 1155–1228. [Google Scholar]
- Nordlund, J.J. The melanocyte and the epidermal melanin unit: An expanded concept. Dermatol. Clin 2007, 25, 271–281. [Google Scholar]
- Slominski, A.; Wortsman, J.; Plonka, P.M.; Schallreuter, K.U.; Paus, R.; Tobin, D.J. Hair follicle pigmentation. J. Invest. Dermatol 2005, 124, 13–21. [Google Scholar]
- Scherer, D.; Kumar, R. Genetics of pigmentation in skin cancer—A review. Mutat. Res 2010, 705, 141–153. [Google Scholar]
- Jimbow, K.; Salopek, T.G.; Dixon, W.T.; Searles, G.E.; Yamada, K. The epidermal melanin unit in the pathophysiology of malignant melanoma. Am. J. Dermatopathol 1991, 13, 179–188. [Google Scholar]
- Joshi, P.G.; Nair, N.; Begum, G.; Joshi, N.B.; Sinkar, V.P.; Vora, S. Melanocyte-keratinocyte interaction induces calcium signalling and melanin transfer to keratinocytes. Pigment. Cell Res 2007, 20, 380–384. [Google Scholar]
- Cardinali, G.; Bolasco, G.; Aspite, N.; Lucania, G.; Lotti, L.V.; Torrisi, M.R.; Picardo, M. Melanosome transfer promoted by keratinocyte growth factor in light and dark skin-derived keratinocytes. J. Invest. Dermatol 2008, 128, 558–567. [Google Scholar]
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. Biofactors 2009, 35, 193–199. [Google Scholar]
- Slominski, A.; Paus, R.; Schadendorf, D. Melanocytes as “sensory” and regulatory cells in the epidermis. J. Theor. Biol 1993, 164, 103–120. [Google Scholar]
- Kalka, K.; Mukhtar, H.; Turowski-Wanke, A.; Merk, H. Biomelanin antioxidants in cosmetics: Assessment based on inhibition of lipid peroxidation. Skin Pharmacol. Appl. Skin Physiol 2000, 13, 143–149. [Google Scholar]
- Mackintosh, J.A. The antimicrobial properties of melanocytes, melanosomes and melanin and the evolution of black skin. J. Theor. Biol 2001, 211, 101–113. [Google Scholar]
- Meyskens, F.L., Jr; Farmer, P.; Fruehauf, J.P. Redox regulation in human melanocytes and melanoma. Pigment. Cell Res 2001, 14, 148–154. [Google Scholar]
- Double, K.L.; Ben-Shachar, D.; Youdim, M.B.; Zecca, L.; Riederer, P.; Gerlach, M. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotoxicol. Teratol 2002, 24, 621–628. [Google Scholar]
- Herrling, T.; Jung, K.; Fuchs, J. The role of melanin as protector against free radicals in skin and its role as free radical indicator in hair. Spectrochim. Acta A Mol. Biomol. Spectrosc 2008, 69, 1429–1435. [Google Scholar]
- Wang, A.; Marino, A.R.; Gasyna, Z.; Gasyna, E.; Norris, J., Jr. Photoprotection by porcine eumelanin against singlet oxygen production. Photochem. PhotoBiol 2008, 84, 679–682. [Google Scholar]
- Riley, P.A. Melanin. Int. J. Biochem. Cell. Biol 1997, 29, 1235–1239. [Google Scholar]
- Meredith, P.; Sarna, T. The physical and chemical properties of eumelanin. Pigment. Cell Res 2006, 19, 572–594. [Google Scholar]
- Slominski, A.; Paus, R. Are l-tyrosine and l-dopa hormone-like bioregulators? J. Theor. Biol 1990, 143, 123–138. [Google Scholar]
- Slominski, A. Neuroendocrine activity of the melanocyte. Exp. Dermatol 2009, 18, 760–763. [Google Scholar]
- Slominski, A.; Zmijewski, M.A.; Pawelek, J. l-tyrosine and l-dihydroxyphenylalanine as hormone-like regulators of melanocyte functions. Pigment. Cell Melanoma Res 2012, 25, 14–27. [Google Scholar]
- Ito, S.; Wakamatsu, K.; Ozeki, H. Chemical analysis of melanins and its application to the study of the regulation of melanogenesis. Pigment. Cell Res 2000, 13, 103–109. [Google Scholar]
- Vincensi, M.R.; d’Ischia, M.; Napolitano, A.; Procaccini, E.M.; Riccio, G.; Monfrecola, G.; Santoianni, P.; Prota, G. Phaeomelanin versus eumelanin as a chemical indicator of ultraviolet sensitivity in fair-skinned subjects at high risk for melanoma: A pilot study. Melanoma Res 1998, 8, 53–58. [Google Scholar]
- Benedetto, J.P.; Ortonne, J.P.; Voulot, C.; Khatchadourian, C.; Prota, G.; Thivolet, J. Role of thiol compounds in mammalian melanin pigmentation: Part I. Reduced and oxidized glutathione. J. Invest. Dermatol 1981, 77, 402–405. [Google Scholar]
- Benedetto, J.P.; Ortonne, J.P.; Voulot, C.; Khatchadourian, C.; Prota, G.; Thivolet, J. Role of thiol compounds in mammalian melanin pigmentation. II. Glutathione and related enzymatic activities. J. Invest. Dermatol 1982, 79, 422–424. [Google Scholar]
- Sealy, R.C.; Hyde, J.S.; Felix, C.C.; Menon, I.A.; Prota, G.; Swartz, H.M.; Persad, S.; Haberman, H.F. Novel free radicals in synthetic and natural pheomelanins: Distinction between dopa melanins and cysteinyldopa melanins by ESR spectroscopy. Proc. Natl. Acad. Sci. USA 1982, 79, 2885–2889. [Google Scholar]
- Costantini, C.; d’Ischia, M.; Palumbo, A.; Prota, G. Photochemistry of 5-S-cysteinyldopa. Photochem. PhotoBiol 1994, 60, 33–37. [Google Scholar]
- Prota, G. Melanins, melanogenesis and melanocytes: Looking at their functional significance from the chemist’s viewpoint. Pigment. Cell Res 2000, 13, 283–293. [Google Scholar]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar]
- Lu, H.; Edwards, C.; Gaskell, S.; Pearse, A.; Marks, R. Melanin content and distribution in the surface corneocyte with skin phototypes. Br. J. Dermatol 1996, 135, 263–267. [Google Scholar]
- Andreassi, L.; Flori, M.L.; Rubegni, P. Sun and skin—Role of phototype and skin colour. Rheumaderm 1999, 455, 469–475. [Google Scholar]
- Kawada, A. Risk and preventive factors for skin phototype. J. Dermatol. Sci 2000, 23, S27–S29. [Google Scholar]
- Ravnbak, M.H. Objective determination of Fitzpatrick skin type. Dan. Med. Bull 2010, 57, B4153. [Google Scholar]
- Godar, D.E. UV doses worldwide. Photochem. PhotoBiol 2005, 81, 736–749. [Google Scholar]
- Dore, J.F.; Chignol, M.C. Tanning salons and skin cancer. Photochem. PhotoBiol. Sci 2012, 11, 30–37. [Google Scholar]
- Elwood, J.M.; Diffey, B.L. A consideration of ambient solar ultraviolet radiation in the interpretation of studies of the aetiology of melanoma. Melanoma Res 1993, 3, 113–122. [Google Scholar]
- Tatalovich, Z.; Wilson, J.P.; Mack, T.; Yan, Y.; Cockburn, M. The objective assessment of lifetime cumulative ultraviolet exposure for determining melanoma risk. J. Photochem. PhotoBiol. B 2006, 85, 198–204. [Google Scholar]
- Qureshi, A.A.; Laden, F.; Colditz, G.A.; Hunter, D.J. Geographic variation and risk of skin cancer in US women. Differences between melanoma, squamous cell carcinoma, and basal cell carcinoma. Arch. Intern. Med 2008, 168, 501–507. [Google Scholar]
- Choi, K.; Lazovich, D.; Southwell, B.; Forster, J.; Rolnick, S.J.; Jackson, J. Prevalence and characteristics of indoor tanning use among men and women in the United States. Arch. Dermatol 2010, 146, 1356–1361. [Google Scholar]
- Hornung, R.L.; Magee, K.H.; Lee, W.J.; Hansen, L.A.; Hsieh, Y.C. Tanning facility use: Are we exceeding food and drug administration limits? J. Am. Acad. Dermatol 2003, 49, 655–661. [Google Scholar]
- Nilsen, L.T.; Aalerud, T.N.; Hannevik, M.; Veierod, M.B. UVB and UVA irradiances from indoor tanning devices. Photochem. PhotoBiol. Sci 2011, 10, 1129–1136. [Google Scholar]
- Nolan, B.V.; Feldman, S.R. Ultraviolet tanning addiction. Dermatol. Clin 2009, 27, 109–112. [Google Scholar]
- Kourosh, A.S.; Harrington, C.R.; Adinoff, B. Tanning as a behavioral addiction. Am. J. Drug Alcohol. Abuse 2010, 36, 284–290. [Google Scholar]
- Harrington, C.R.; Beswick, T.C.; Leitenberger, J.; Minhajuddin, A.; Jacobe, H.T.; Adinoff, B. Addictive-like behaviours to ultraviolet light among frequent indoor tanners. Clin. Exp. Dermatol 2011, 36, 33–38. [Google Scholar]
- Poorsattar, S.P.; Hornung, R.L. UV light abuse and high-risk tanning behavior among undergraduate college students. J. Am. Acad. Dermatol 2007, 56, 375–379. [Google Scholar]
- Karagas, M.R.; Stannard, V.A.; Mott, L.A.; Slattery, M.J.; Spencer, S.K.; Weinstock, M.A. Use of tanning devices and risk of basal cell and squamous cell skin cancers. J. Natl. Cancer Inst 2002, 94, 224–226. [Google Scholar]
- Levine, J.A.; Sorace, M.; Spencer, J.; Siegel, D.M. The indoor UV tanning industry: A review of skin cancer risk, health benefit claims, and regulation. J. Am. Acad. Dermatol 2005, 53, 1038–1044. [Google Scholar]
- Schulman, J.M.; Fisher, D.E. Indoor ultraviolet tanning and skin cancer: Health risks and opportunities. Curr. Opin. Oncol 2009, 21, 144–149. [Google Scholar]
- Fisher, D.E.; James, W.D. Indoor tanning—Science, behavior, and policy. N. Eng. J. Med 2010, 363, 901–903. [Google Scholar]
- Weinstock, M.A.; Fisher, D.E. Indoor ultraviolet tanning: What the data do and do not show regarding risk of melanoma and keratinocyte malignancies. J. Natl. Compr. Canc. Netw 2010, 8, 867–873. [Google Scholar]
- Han, J.; Colditz, G.A.; Hunter, D.J. Risk factors for skin cancers: A nested case-control study within the Nurses’ Health Study. Int. J. Epidemiol 2006, 35, 1514–1521. [Google Scholar]
- Lazovich, D.; Vogel, R.I.; Berwick, M.; Weinstock, M.A.; Anderson, K.E.; Warshaw, E.M. Indoor tanning and risk of melanoma: A case-control study in a highly exposed population. Cancer Epidemiol. Biomark. Prev 2010, 19, 1557–1568. [Google Scholar]
- Holick, M.F. Sunlight “D”ilemma: Risk of skin cancer or bone disease and muscle weakness. Lancet 2001, 357, 4–6. [Google Scholar]
- Holick, M. Does sunscreen block the skin’s ability to make vitamin D? If so, how can I get enough of this vitamin without raising my risk of skin cancer? Health News 2002, 8, 12. [Google Scholar]
- Tangpricha, V.; Turner, A.; Spina, C.; Decastro, S.; Chen, T.C.; Holick, M.F. Tanning is associated with optimal vitamin D status (serum 25-hydroxyvitamin D concentration) and higher bone mineral density. Am. J. Clin. Nutr 2004, 80, 1645–1649. [Google Scholar]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr 2004, 80, 1678S–1688S. [Google Scholar]
- Rajakumar, K.; Greenspan, S.L.; Thomas, S.B.; Holick, M.F. SOLAR ultraviolet radiation and vitamin D: A historical perspective. Am. J. Public Health 2007, 97, 1746–1754. [Google Scholar]
- Holick, M.F.; Chen, T.C.; Lu, Z.; Sauter, E. Vitamin D and skin physiology: A D-lightful story. J. Bone Miner. Res 2007, 22, V28–V33. [Google Scholar]
- Holick, M.F. Sunlight, UV-radiation, vitamin D and skin cancer: How much sunlight do we need? Adv. Exp. Med. Biol 2008, 624, 1–15. [Google Scholar]
- Holick, M.F. Vitamin D and sunlight: Strategies for cancer prevention and other health benefits. Clin. J. Am. Soc. Nephrol 2008, 3, 1548–1554. [Google Scholar]
- Carbone, L.D.; Rosenberg, E.W.; Tolley, E.A.; Holick, M.F.; Hughes, T.A.; Watsky, M.A.; Barrow, K.D.; Chen, T.C.; Wilkin, N.K.; Bhattacharya, S.K.; et al. 25-Hydroxyvitamin D, cholesterol, and ultraviolet irradiation. Metabolism 2008, 57, 741–748. [Google Scholar]
- Holick, M.F. Vitamin D: A millenium perspective. J. Cell. Biochem 2003, 88, 296–307. [Google Scholar]
- Holick, M.F. Resurrection of vitamin D deficiency and rickets. J. Clin. Investig 2006, 116, 2062–2072. [Google Scholar]
- Holick, M.F.; Chen, T.C. Vitamin D deficiency: A worldwide problem with health consequences. Am. J. Clin. Nutr 2008, 87, 1080S–1086S. [Google Scholar]
- Reichrath, J.; Nurnberg, B. Cutaneous vitamin D synthesis versus skin cancer development: The Janus faces of solar UV-radiation. Dermatoendocrinology 2009, 1, 253–261. [Google Scholar]
- Gallagher, R.P.; Lee, T.K.; Bajdik, C.D.; Borugian, M. Ultraviolet radiation. Chronic. Dis. Can 2010, 29, 51–68. [Google Scholar]
- Lucas, R.M.; McMichael, A.J.; Armstrong, B.K.; Smith, W.T. Estimating the global disease burden due to ultraviolet radiation exposure. Int. J. Epidemiol 2008, 37, 654–667. [Google Scholar]
- Clydesdale, G.J.; Dandie, G.W.; Muller, H.K. Ultraviolet light induced injury: Immunological and inflammatory effects. Immunol. Cell. Biol 2001, 79, 547–568. [Google Scholar]
- Matsumura, Y.; Ananthaswamy, H.N. Toxic effects of ultraviolet radiation on the skin. Toxicol. Appl. Pharmacol 2004, 195, 298–308. [Google Scholar]
- Skobowiat, C.; Dowdy, J.C.; Sayre, R.M.; Tuckey, R.C.; Slominski, A. Cutaneous hypothalamic-pituitary-adrenal axis homolog: Regulation by ultraviolet radiation. Am. J. Physiol. Endocrinol. Metab 2011, 301, E484–E493. [Google Scholar]
- Skobowiat, C.; Sayre, R.M.; Dowdy, J.C.; Slominski, A.T. Ultraviolet radiation regulates cortisol activity in a waveband-dependent manner in human skin ex vivo. Br. J. Dermatol 2013, 168, 595–601. [Google Scholar]
- Bayerl, C.; Taake, S.; Moll, I.; Jung, E.G. Characterization of sunburn cells after exposure to ultraviolet light. PhotoDermatol. Photoimmunol. Photomed 1995, 11, 149–154. [Google Scholar]
- Coelho, S.G.; Choi, W.; Brenner, M.; Miyamura, Y.; Yamaguchi, Y.; Wolber, R.; Smuda, C.; Batzer, J.; Kolbe, L.; Ito, S.; et al. Short- and long-term effects of UV radiation on the pigmentation of human skin. J. Investig. Dermatol. Symp. Proc 2009, 14, 32–35. [Google Scholar]
- Scott, T.L.; Christian, P.A.; Kesler, M.V.; Donohue, K.M.; Shelton, B.; Wakamatsu, K.; Ito, S.; D’Orazio, J. Pigment-independent cAMP-mediated epidermal thickening protects against cutaneous UV injury by keratinocyte proliferation. Exp. Dermatol 2012, 21, 771–777. [Google Scholar]
- Slominski, A.; Wortsman, J.; Pisarchik, A.; Zbytek, B.; Linton, E.A.; Mazurkiewicz, J.E.; Wei, E.T. Cutaneous expression of corticotropin-releasing hormone (CRH), urocortin, and CRH receptors. FASEB J 2001, 15, 1678–1693. [Google Scholar]
- D’Orazio, J.A.; Nobuhisa, T.; Cui, R.; Arya, M.; Spry, M.; Wakamatsu, K.; Igras, V.; Kunisada, T.; Granter, S.R.; Nishimura, E.K.; et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 2006, 443, 340–344. [Google Scholar]
- Slominski, A.; Wortsman, J.; Tuckey, R.C.; Paus, R. Differential expression of HPA axis homolog in the skin. Mol. Cell. Endocrinol. 2007, 265–266, 143–149. [Google Scholar]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central role of p53 in the Suntan response and pathologic hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar]
- McGill, G.G.; Horstmann, M.; Widlund, H.R.; Du, J.; Motyckova, G.; Nishimura, E.K.; Lin, Y.L.; Ramaswamy, S.; Avery, W.; Ding, H.F.; et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 2002, 109, 707–718. [Google Scholar]
- Widlund, H.R.; Fisher, D.E. Microphthalamia-associated transcription factor: A critical regulator of pigment cell development and survival. Oncogene 2003, 22, 3035–3041. [Google Scholar]
- Rouzaud, F.; Costin, G.E.; Yamaguchi, Y.; Valencia, J.C.; Berens, W.F.; Chen, K.G.; Hoashi, T.; Bohm, M.; Abdel-Malek, Z.A.; Hearing, V.J. Regulation of constitutive and UVR-induced skin pigmentation by melanocortin 1 receptor isoforms. FASEB J 2006, 20, 1927–1929. [Google Scholar]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med 2006, 12, 406–414. [Google Scholar]
- Mitra, D.; Fisher, D.E. Transcriptional regulation in melanoma. Hematol. Oncol. Clin. N. Am 2009, 23, 447–465. [Google Scholar]
- Park, S.B.; Huh, C.H.; Choe, Y.B.; Youn, J.I. Time course of ultraviolet-induced skin reactions evaluated by two different reflectance spectrophotometers: DermaSpectrophotometer and Minolta spectrophotometer CM-2002. PhotoDermatol. Photoimmunol. Photomed 2002, 18, 23–28. [Google Scholar]
- Beattie, P.E.; Dawe, R.S.; Ferguson, J.; Ibbotson, S.H. Dose-response and time-course characteristics of UV-A1 erythema. Arch. Dermatol 2005, 141, 1549–1555. [Google Scholar]
- Chedekel, M.R.; Zeise, L. Sunlight, melanogenesis and radicals in the skin. Lipids 1988, 23, 587–591. [Google Scholar]
- Pawelek, J.M.; Chakraborty, A.K.; Osber, M.P.; Orlow, S.J.; Min, K.K.; Rosenzweig, K.E.; Bolognia, J.L. Molecular cascades in UV-induced melanogenesis: A central role for melanotropins? Pigment. Cell Res 1992, 5, 348–356. [Google Scholar]
- Eller, M.S.; Ostrom, K.; Gilchrest, B.A. DNA damage enhances melanogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 1087–1092. [Google Scholar]
- Gilchrest, B.A.; Park, H.Y.; Eller, M.S.; Yaar, M. Mechanisms of ultraviolet light-induced pigmentation. Photochem. PhotoBiol 1996, 63, 1–10. [Google Scholar]
- Funasaka, Y.; Chakraborty, A.K.; Hayashi, Y.; Komoto, M.; Ohashi, A.; Nagahama, M.; Inoue, Y.; Pawelek, J.; Ichihashi, M. Modulation of melanocyte-stimulating hormone receptor expression on normal human melanocytes: Evidence for a regulatory role of ultraviolet B, interleukin-1alpha, interleukin-1beta, endothelin-1 and tumour necrosis factor-alpha. Br. J. Dermatol 1998, 139, 216–224. [Google Scholar]
- Duval, C.; Regnier, M.; Schmidt, R. Distinct melanogenic response of human melanocytes in mono-culture, in co-culture with keratinocytes and in reconstructed epidermis, to UV exposure. Pigment. Cell Res 2001, 14, 348–355. [Google Scholar]
- Raffin-Sanson, M.L.; de Keyzer, Y.; Bertagna, X. Proopiomelanocortin, a polypeptide precursor with multiple functions: From physiology to pathological conditions. Eur. J. Endocrinol 2003, 149, 79–90. [Google Scholar]
- Corre, S.; Primot, A.; Sviderskaya, E.; Bennett, D.C.; Vaulont, S.; Goding, C.R.; Galibert, M.D. UV-induced expression of key component of the tanning process, the POMC and MC1R genes, is dependent on the p-38-activated upstream stimulating factor-1 (USF-1). J. Biol. Chem 2004, 279, 51226–51233. [Google Scholar]
- Slominski, A.; Tobin, D.J.; Paus, R. Does p53 regulate skin pigmentation by controlling proopiomelanocortin gene transcription? Pigment. Cell Res 2007, 20, 307–308. [Google Scholar]
- Kripke, M.L.; Fisher, M.S. Immunologic parameters of ultraviolet carcinogenesis. J. Natl. Cancer Inst 1976, 57, 211–215. [Google Scholar]
- Kripke, M.L.; Lofgreen, J.S.; Beard, J.; Jessup, J.M.; Fisher, M.S. In vivo immune responses of mice during carcinogenesis by ultraviolet irradiation. J. Natl. Cancer Inst 1977, 59, 1227–1230. [Google Scholar]
- Kripke, M.L. Effects of UV radiation on the immune system: Consequences for UV carcinogenesis. Adv. Exp. Med. Biol 1979, 121, 589–598. [Google Scholar]
- Kripke, M.L. Immunological unresponsiveness induced by ultraviolet radiation. Immunol. Rev 1984, 80, 87–102. [Google Scholar]
- Schade, N.; Esser, C.; Krutmann, J. Ultraviolet B radiation-induced immunosuppression: Molecular mechanisms and cellular alterations. Photochem. PhotoBiol. Sci 2005, 4, 699–708. [Google Scholar]
- Norval, M. The mechanisms and consequences of ultraviolet-induced immunosuppression. Prog. Biophys. Mol. Biol 2006, 92, 108–118. [Google Scholar]
- De Gruijl, F.R. UV-induced immunosuppression in the balance. Photochem. PhotoBiol 2008, 84, 2–9. [Google Scholar]
- Kripke, M.L. Reflections on the field of photoimmunology. J. Invest. Dermatol 2013, 133, 27–30. [Google Scholar]
- Polefka, T.G.; Meyer, T.A.; Agin, P.P.; Bianchini, R.J. Effects of solar radiation on the skin. J. Cosmet. Dermatol 2012, 11, 134–143. [Google Scholar]
- Sklar, L.R.; Almutawa, F.; Lim, H.W.; Hamzavi, I. Effects of ultraviolet radiation, visible light, and infrared radiation on erythema and pigmentation: A review. Photochem. PhotoBiol. Sci 2013, 12, 54–64. [Google Scholar]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res 2005, 571, 19–31. [Google Scholar]
- Tyrrell, R.M. Ultraviolet radiation and free radical damage to skin. Biochem. Soc. Symp 1995, 61, 47–53. [Google Scholar]
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol 2004, 43, 326–335. [Google Scholar]
- Burke, K.E. Photoaging: The role of oxidative stress. G Ital. Dermatol. Venereol 2010, 145, 445–459. [Google Scholar]
- De Gruijl, F.R. Photocarcinogenesis: UVA vs. UVB. Methods Enzymol 2000, 319, 359–366. [Google Scholar]
- Ikehata, H.; Ono, T. The mechanisms of UV mutagenesis. J. Radiat. Res 2011, 52, 115–125. [Google Scholar]
- Sage, E.; Girard, P.M.; Francesconi, S. Unravelling UVA-induced mutagenesis. Photochem. PhotoBiol. Sci 2012, 11, 74–80. [Google Scholar]
- Schulz, I.; Mahler, H.C.; Boiteux, S.; Epe, B. Oxidative DNA base damage induced by singlet oxygen and photosensitization: Recognition by repair endonucleases and mutagenicity. Mutat. Res 2000, 461, 145–156. [Google Scholar]
- Nishimura, S. Involvement of mammalian OGG1(MMH) in excision of the 8-hydroxyguanine residue in DNA. Free Radic. Biol. Med 2002, 32, 813–821. [Google Scholar]
- Kunisada, M.; Sakumi, K.; Tominaga, Y.; Budiyanto, A.; Ueda, M.; Ichihashi, M.; Nakabeppu, Y.; Nishigori, C. 8-Oxoguanine formation induced by chronic UVB exposure makes Ogg1 knockout mice susceptible to skin carcinogenesis. Cancer Res 2005, 65, 6006–6010. [Google Scholar]
- Agar, N.S.; Halliday, G.M.; Barnetson, R.S.; Ananthaswamy, H.N.; Wheeler, M.; Jones, A.M. The basal layer in human squamous tumors harbors more UVA than UVB fingerprInt. mutations: A role for UVA in human skin carcinogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 4954–4959. [Google Scholar]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Gaze, D.C.; Tobin, D.J.; Marshall, H.S.; Panske, A.; Panzig, E.; Hibberts, N.A. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J. Investig. Dermatol. Symp. Proc 1999, 4, 91–96. [Google Scholar]
- Song, X.; Mosby, N.; Yang, J.; Xu, A.; Abdel-Malek, Z.; Kadekaro, A.L. Alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment. Cell Melanoma Res 2009, 22, 809–818. [Google Scholar]
- Kadekaro, A.L.; Chen, J.; Yang, J.; Chen, S.; Jameson, J.; Swope, V.B.; Cheng, T.; Kadakia, M.; Abdel-Malek, Z. Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol. Cancer Res 2012, 10, 778–786. [Google Scholar]
- Krol, E.S.; Kramer-Stickland, K.A.; Liebler, D.C. Photoprotective actions of topically applied vitamin E. Drug Metab. Rev 2000, 32, 413–420. [Google Scholar]
- Bickers, D.R.; Athar, M. Oxidative stress in the pathogenesis of skin disease. J. Invest. Dermatol 2006, 126, 2565–2575. [Google Scholar]
- Kokot, A.; Metze, D.; Mouchet, N.; Galibert, M.D.; Schiller, M.; Luger, T.A.; Bohm, M. Alpha-melanocyte-stimulating hormone counteracts the suppressive effect of UVB on Nrf2 and Nrf-dependent gene expression in human skin. Endocrinology 2009, 150, 3197–206. [Google Scholar]
- Cleaver, J.E.; Crowley, E. UV damage, DNA repair and skin carcinogenesis. Front. BioSci 2002, 7, d1024–d1043. [Google Scholar]
- Wei, Q.; Lee, J.E.; Gershenwald, J.E.; Ross, M.I.; Mansfield, P.F.; Strom, S.S.; Wang, L.E.; Guo, Z.; Qiao, Y.; Amos, C.I.; et al. Repair of UV light-induced DNA damage and risk of cutaneous malignant melanoma. J. Natl. Cancer Inst 2003, 95, 308–315. [Google Scholar]
- Sarasin, A. The molecular pathways of ultraviolet-induced carcinogenesis. Mutat. Res 1999, 428, 5–10. [Google Scholar]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Eng. J. Med 2009, 361, 1475–1485. [Google Scholar]
- Kanjilal, S.; Pierceall, W.E.; Cummings, K.K.; Kripke, M.L.; Ananthaswamy, H.N. High frequency of p53 mutations in ultraviolet radiation-induced murine skin tumors: Evidence for strand bias and tumor heterogeneity. Cancer Res 1993, 53, 2961–2964. [Google Scholar]
- Sato, M.; Nishigori, C.; Zghal, M.; Yagi, T.; Takebe, H. Ultraviolet-specific mutations in p53 gene in skin tumors in xeroderma pigmentosum patients. Cancer Res 1993, 53, 2944–2946. [Google Scholar]
- Daya-Grosjean, L.; Dumaz, N.; Sarasin, A. The specificity of p53 mutation spectra in sunlight induced human cancers. J. Photochem. PhotoBiol. B 1995, 28, 115–124. [Google Scholar]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci 2009, 66, 994–1009. [Google Scholar]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on xeroderma pigmentosum. J. Invest. Dermatol 2012, 132, 785–796. [Google Scholar]
- Daya-Grosjean, L. Xeroderma pigmentosum and skin cancer. Adv. Exp. Med. Biol 2008, 637, 19–27. [Google Scholar]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Ann. Rev. Biochem 2004, 73, 39–85. [Google Scholar]
- Reed, S.H. Nucleotide excision repair in chromatin: The shape of things to come. DNA Repair 2005, 4, 909–918. [Google Scholar]
- Leibeling, D.; Laspe, P.; Emmert, S. Nucleotide excision repair and cancer. J. Mol. Histol 2006, 37, 225–238. [Google Scholar]
- Rogers, H.W.; Weinstock, M.A.; Harris, A.R.; Hinckley, M.R.; Feldman, S.R.; Fleischer, A.B.; Coldiron, B.M. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch. Dermatol 2010, 146, 283–287. [Google Scholar]
- Donaldson, M.R.; Coldiron, B.M. No end in sight: The skin cancer epidemic continues. Semin. Cutan. Med. Surg 2011, 30, 3–5. [Google Scholar]
- De Gruijl, F.R. Skin cancer and solar UV radiation. Eur. J. Cancer 1999, 35, 2003–2009. [Google Scholar]
- Chen, J.G.; Fleischer, A.B., Jr; Smith, E.D.; Kancler, C.; Goldman, N.D.; Williford, P.M.; Feldman, S.R. Cost of nonmelanoma skin cancer treatment in the United States. Dermatol. Surg 2001, 27, 1035–1038. [Google Scholar]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol 2010, 49, 978–986. [Google Scholar]
- Berwick, M.; Wiggins, C. The current epidemiology of cutaneous malignant melanoma. Front. BioSci 2006, 11, 1244–1254. [Google Scholar]
- Croyle, R.T. SEER Stat Fact Sheets: Melanoma of the Skin. Available online: http://www.seer.cancer.gov/statfacts/html/melan.html#incidence-mortality (accessed on 4 March 2013).
- Marks, R. Epidemiology of melanoma. Clin. Exp. Dermatol 2000, 25, 459–463. [Google Scholar]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Eng. J. Med 2010, 363, 809–819. [Google Scholar]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Eng. J. Med 2012, 366, 707–714. [Google Scholar]
- Nikolaou, V.A.; Stratigos, A.J.; Flaherty, K.T.; Tsao, H. Melanoma: New insights and new therapies. J. Invest. Dermatol 2012, 132, 854–863. [Google Scholar]
- Ji, Z.; Flaherty, K.T.; Tsao, H. Targeting the RAS pathway in melanoma. Trends Mol. Med 2012, 18, 27–35. [Google Scholar]
- Flaherty, K.T. Targeting metastatic melanoma. Annu. Rev. Med 2012, 63, 171–183. [Google Scholar]
- Hodi, F.S.; Oble, D.A.; Drappatz, J.; Velazquez, E.F.; Ramaiya, N.; Ramakrishna, N.; Day, A.L.; Kruse, A.; Mac Rae, S.; Hoos, A.; et al. CTLA-4 blockade with ipilimumab induces significant clinical benefit in a female with melanoma metastases to the CNS. Nat. Clin. Pract. Oncol 2008, 5, 557–561. [Google Scholar]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Eng. J. Med 2010, 363, 711–723. [Google Scholar]
- Franceschi, S.; Cristofolini, M. Cutaneous malignant melanoma: Epidemiological considerations. Semin. Surg. Oncol 1992, 8, 345–352. [Google Scholar]
- Rivers, J.K. The detection and management of dysplastic nevi and early melanoma. World J. Surg 1992, 16, 166–172. [Google Scholar]
- Garland, C.F.; Garland, F.C.; Gorham, E.D. Rising trends in melanoma. An hypothesis concerning sunscreen effectiveness. Ann. Epidemiol 1993, 3, 103–110. [Google Scholar]
- Boyle, P.; Maisonneuve, P.; Dore, J.F. Epidemiology of malignant melanoma. Br. Med. Bull 1995, 51, 523–547. [Google Scholar]
- Liu, T.; Soong, S.J. Epidemiology of malignant melanoma. Surg. Clin. N. Am 1996, 76, 1205–1222. [Google Scholar]
- Weinstock, M.A. Issues in the epidemiology of melanoma. Hematol. Oncol. Clin. N. Am 1998, 12, 681–698. [Google Scholar]
- Brochez, L.; Naeyaert, J.M. Understanding the trends in melanoma incidence and mortality: Where do we stand? Eur. J. Dermatol 2000, 10, 71–75. [Google Scholar]
- Diepgen, T.L.; Mahler, V. The epidemiology of skin cancer. Br. J. Dermatol 2002, 146, 1–6. [Google Scholar]
- Rigel, D.S. The effect of sunscreen on melanoma risk. Dermatol. Clin 2002, 20, 601–606. [Google Scholar]
- Weinstock, M.A. Cutaneous melanoma: Public health approach to early detection. Dermatol. Ther 2006, 19, 26–31. [Google Scholar]
- Norval, M.; Cullen, A.P.; de Gruijl, F.R.; Longstreth, J.; Takizawa, Y.; Lucas, R.M.; Noonan, F.P.; van der Leun, J.C. The effects on human health from stratospheric ozone depletion and its interactions with climate change. Photochem. PhotoBiol. Sci 2007, 6, 232–251. [Google Scholar]
- Leiter, U.; Garbe, C. Epidemiology of melanoma and nonmelanoma skin cancer—The role of sunlight. Adv. Exp. Med. Biol 2008, 624, 89–103. [Google Scholar]
- Tucker, M.A. Melanoma epidemiology. Hematol. Oncol. Clin. N. Am 2009, 23, 383–395. [Google Scholar]
- Norval, M.; Lucas, R.M.; Cullen, A.P.; de Gruijl, F.R.; Longstreth, J.; Takizawa, Y.; van der Leun, J.C. The human health effects of ozone depletion and interactions with climate change. Photochem. PhotoBiol. Sci 2011, 10, 199–225. [Google Scholar]
- Linos, E.; Swetter, S.M.; Cockburn, M.G.; Colditz, G.A.; Clarke, C.A. Increasing burden of melanoma in the United States. J. Invest. Dermatol 2009, 129, 1666–1674. [Google Scholar]
- Armstrong, B.K.; Kricker, A. How much melanoma is caused by sun exposure? Melanoma Res 1993, 3, 395–401. [Google Scholar]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordonez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar]
- Ling, G.; Persson, A.; Berne, B.; Uhlen, M.; Lundeberg, J.; Ponten, F. Persistent p53 mutations in single cells from normal human skin. Am. J. Pathol 2001, 159, 1247–1253. [Google Scholar]
- Lacour, J.P. Carcinogenesis of basal cell carcinomas: Genetics and molecular mechanisms. Br. J. Dermatol 2002, 146, 17–19. [Google Scholar]
- Bolshakov, S.; Walker, C.M.; Strom, S.S.; Selvan, M.S.; Clayman, G.L.; El-Naggar, A.; Lippman, S.M.; Kripke, M.L.; Ananthaswamy, H.N. p53 mutations in human aggressive and nonaggressive basal and squamous cell carcinomas. Clin. Cancer Res 2003, 9, 228–234. [Google Scholar]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol 2005, 152, 43–51. [Google Scholar]
- Daya-Grosjean, L.; Sarasin, A. The role of UV induced lesions in skin carcinogenesis: An overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat. Res 2005, 571, 43–56. [Google Scholar]
- Queille, S.; Luron, L.; Spatz, A.; Avril, M.F.; Ribrag, V.; Duvillard, P.; Hiesse, C.; Sarasin, A.; Armand, J.P.; Daya-Grosjean, L. Analysis of skin cancer risk factors in immunosuppressed renal transplant patients shows high levels of UV-specific tandem CC to TT mutations of the p53 gene. Carcinogenesis 2007, 28, 724–731. [Google Scholar]
- Benjamin, C.L.; Melnikova, V.O.; Ananthaswamy, H.N. P53 protein and pathogenesis of melanoma and nonmelanoma skin cancer. Adv. Exp. Med. Biol 2008, 624, 265–282. [Google Scholar]
- Beissert, S.; Loser, K. Molecular and cellular mechanisms of photocarcinogenesis. Photochem. PhotoBiol 2008, 84, 29–34. [Google Scholar]
- Rees, J.L. The genetics of sun sensitivity in humans. Am. J. Hum. Genet 2004, 75, 739–751. [Google Scholar]
- Valverde, P.; Healy, E.; Jackson, I.; Rees, J.L.; Thody, A.J. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat. Genet 1995, 11, 328–330. [Google Scholar]
- Valverde, P.; Healy, E.; Sikkink, S.; Haldane, F.; Thody, A.J.; Carothers, A.; Jackson, I.J.; Rees, J.L. The Asp84Glu variant of the melanocortin 1 receptor (MC1R) is associated with melanoma. Hum. Mol. Genet 1996, 5, 1663–1666. [Google Scholar]
- Koppula, S.V.; Robbins, L.S.; Lu, D.; Baack, E.; White, C.R., Jr; Swanson, N.A.; Cone, R.D. Identification of common polymorphisms in the coding sequence of the human MSH receptor (MCIR) with possible biological effects. Hum. Mutat 1997, 9, 30–36. [Google Scholar]
- Rees, J.L.; Healy, E. Melanocortin receptors, red hair, and skin cancer. J. Investig. Dermatol. Symp. Proc 1997, 2, 94–98. [Google Scholar]
- Abdel-Malek, Z.A.; Kadekaro, A.L.; Kavanagh, R.J.; Todorovic, A.; Koikov, L.N.; McNulty, J.C.; Jackson, P.J.; Millhauser, G.L.; Schwemberger, S.; Babcock, G.; et al. Melanoma prevention strategy based on using tetrapeptide alpha-MSH analogs that protect human melanocytes from UV-induced DNA damage and cytotoxicity. FASEB J 2006, 20, 1561–1563. [Google Scholar]
- Landi, M.T.; Bauer, J.; Pfeiffer, R.M.; Elder, D.E.; Hulley, B.; Minghetti, P.; Calista, D.; Kanetsky, P.A.; Pinkel, D.; Bastian, B.C. MC1R germline variants confer risk for BRAF-mutant melanoma. Science Signal 2006, 313, 521. [Google Scholar]
- Roberts, D.W.; Newton, R.A.; Leonard, J.H.; Sturm, R.A. Melanocytes expressing MC1R polymorphisms associated with red hair color have altered MSH-ligand activated pigmentary responses in coculture with keratinocytes. J. Cell. Physiol 2008, 215, 344–355. [Google Scholar]
- Abdel-Malek, Z.A.; Ruwe, A.; Kavanagh-Starner, R.; Kadekaro, A.L.; Swope, V.; Haskell-Luevano, C.; Koikov, L.; Knittel, J.J. Alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigment. Cell Melanoma Res 2009, 22, 635–644. [Google Scholar]
- Suzuki, I.; Cone, R.D.; Im, S.; Nordlund, J.; Abdel-Malek, Z.A. Binding of melanotropic hormones to the melanocortin receptor MC1R on human melanocytes stimulates proliferation and melanogenesis. Endocrinology 1996, 137, 1627–1633. [Google Scholar]
- Sturm, R.A.; Duffy, D.L.; Box, N.F.; Newton, R.A.; Shepherd, A.G.; Chen, W.; Marks, L.H.; Leonard, J.H.; Martin, N.G. Genetic association and cellular function of MC1R variant alleles in human pigmentation. Ann. N. Y. Acad. Sci 2003, 994, 348–358. [Google Scholar]
- Kadekaro, A.L.; Kanto, H.; Kavanagh, R.; Abdel-Malek, Z.A. Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann. N. Y. Acad. Sci 2003, 994, 359–365. [Google Scholar]
- Bertolotto, C.; Abbe, P.; Hemesath, T.J.; Bille, K.; Fisher, D.E.; Ortonne, J.P.; Ballotti, R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J. Cell. Biol 1998, 142, 827–835. [Google Scholar]
- Newton, R.A.; Roberts, D.W.; Leonard, J.H.; Sturm, R.A. Human melanocytes expressing MC1R variant alleles show impaired activation of multiple signaling pathways. Peptides 2007, 28, 2387–2396. [Google Scholar]
- Hauser, J.E.; Kadekaro, A.L.; Kavanagh, R.J.; Wakamatsu, K.; Terzieva, S.; Schwemberger, S.; Babcock, G.; Rao, M.B.; Ito, S.; Abdel-Malek, Z.A. Melanin content and MC1R function independently affect UVR-induced DNA damage in cultured human melanocytes. Pigment. Cell Res 2006, 19, 303–314. [Google Scholar]
- Sturm, R.A. Skin colour and skin cancer—MC1R, the genetic link. Melanoma Res 2002, 12, 405–416. [Google Scholar]
- Beaumont, K.A.; Shekar, S.N.; Cook, A.L.; Duffy, D.L.; Sturm, R.A. Red hair is the null phenotype of MC1R. Hum. Mutat 2008, 29, E88–E94. [Google Scholar]
- Landi, M.T.; Kanetsky, P.A.; Tsang, S.; Gold, B.; Munroe, D.; Rebbeck, T.; Swoyer, J.; Ter-Minassian, M.; Hedayati, M.; Grossman, L.; et al. MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J. Natl. Cancer Inst 2005, 97, 998–1007. [Google Scholar]
- Fargnoli, M.C.; Spica, T.; Sera, F.; Pellacani, G.; Chiarugi, A.; Seidenari, S.; Carli, P.; Chimenti, S.; Peris, K. Re: MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J. Natl. Cancer Inst 2006, 98, 144–145. [Google Scholar]
- Wong, S.S.; Ainger, S.A.; Leonard, J.H.; Sturm, R.A. MC1R variant allele effects on UVR-induced phosphorylation of p38, p53, and DDB2 repair protein responses in melanocytic cells in culture. J. Invest. Dermatol 2012, 132, 1452–1461. [Google Scholar]
- Slominski, A.; Plonka, P.M.; Pisarchik, A.; Smart, J.L.; Tolle, V.; Wortsman, J.; Low, M.J. Preservation of eumelanin hair pigmentation in proopiomelanocortin-deficient mice on a nonagouti (a/a) genetic background. Endocrinology 2005, 146, 1245–1253. [Google Scholar]
- Spry, M.L.; Vanover, J.C.; Scott, T.; Abona-Ama, O.; Wakamatsu, K.; Ito, S.; D’Orazio, J.A. Prolonged treatment of fair-skinned mice with topical forskolin causes persistent tanning and UV protection. Pigment. Cell Melanoma Res 2009, 22, 219–229. [Google Scholar]
- Khaled, M.; Levy, C.; Fisher, D.E. Control of melanocyte differentiation by a MITF-PDE4D3 homeostatic circuit. Genes Dev 2010, 24, 2276–2281. [Google Scholar]
- Ruwe, A.R.; Koikov, L.; Abdel-Malek, Z.; Haskell-Luevano, C.; Dirain, M.L.; Portillo, F.; Xiang, Z.; Wortman, M.; Knittel, J.J. Semi-rigid tripeptide agonists of melanocortin receptors. Bioorg. Med. Chem. Lett 2009, 19, 5176–5181. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
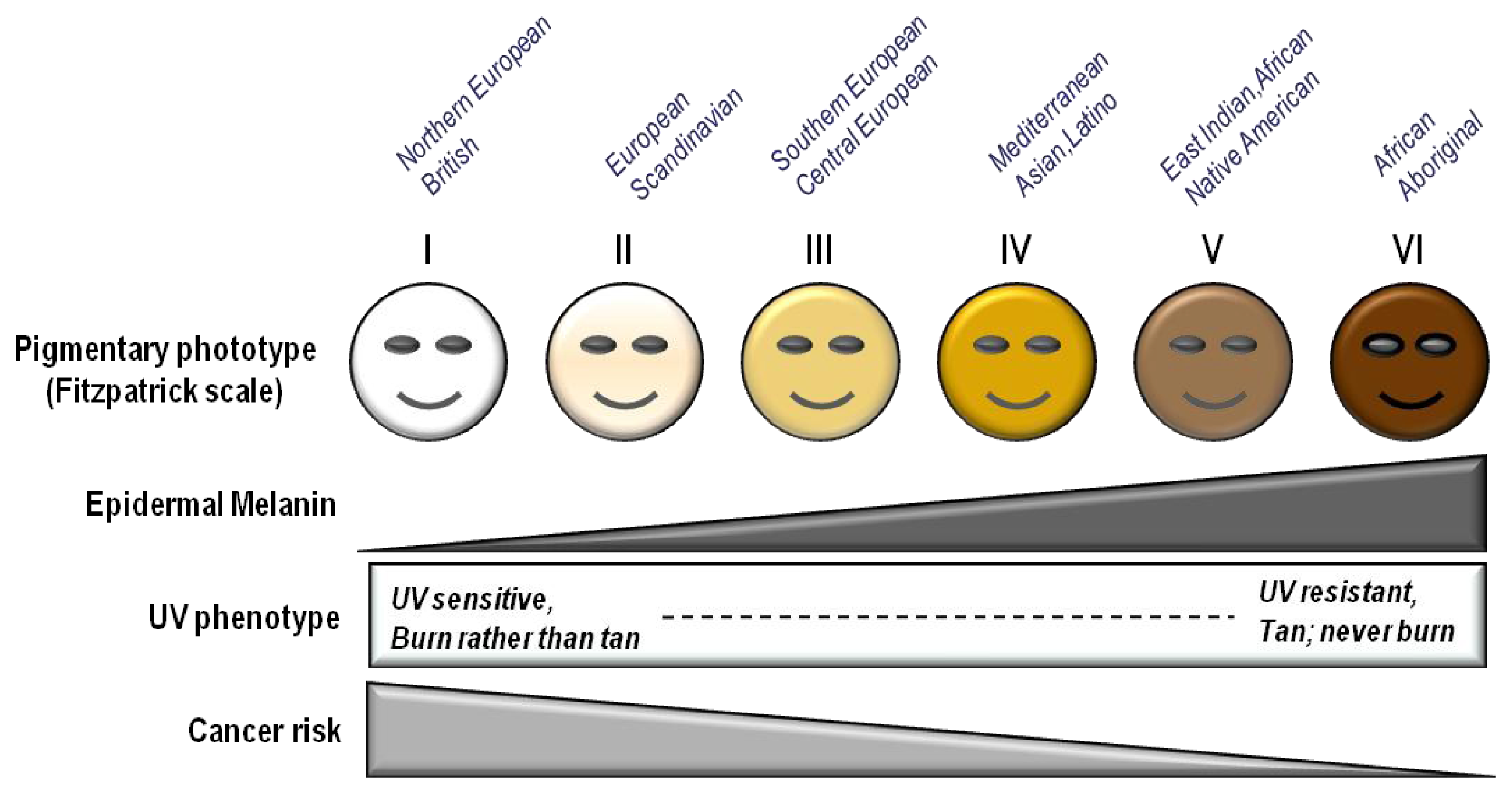
| Fitzpatrick phototype | Phenotype | Epidermal eumelanin | Cutaneous response to UV | MED (mJ/cm2) * | Cancer risk |
|---|---|---|---|---|---|
| I | Unexposed skin is bright white Blue/green eyes typical Freckling frequent Northern European/British | +/− | Always burns Peels Never tans | 15–30 | ++++ |
| II | Unexposed skin is white Blue, hazel or brown eyes Red, blonde or brown hair European/Scandinavian | + | Burns easily Peels Tans minimally | 25–40 | +++/++++ |
| III | Unexposed skin is fair Brown eyes Dark hair Southern or Central European | ++ | Burns moderately Average tanning ability | 30–50 | +++ |
| IV | Unexposed skin is light brown Dark eyes Dark hair Mediterranean, Asian or Latino | +++ | Burns minimally Tans easily | 40–60 | ++ |
| V | Unexposed skin is brown Dark eyes Dark hair East Indian, Native American, Latino or African | ++++ | Rarely burns Tans easily and substantially | 60–90 | + |
| VI | Unexposed skin is black Dark eyes Dark hair African or Aboriginal ancestry | +++++ | Almost never burns Tans readily and profusely | 90–150 | +/− |
| Sun exposure |
|
| Artificial Tanning |
|
| Awareness |
|
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D'Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV Radiation and the Skin. Int. J. Mol. Sci. 2013, 14, 12222-12248. https://doi.org/10.3390/ijms140612222
D'Orazio J, Jarrett S, Amaro-Ortiz A, Scott T. UV Radiation and the Skin. International Journal of Molecular Sciences. 2013; 14(6):12222-12248. https://doi.org/10.3390/ijms140612222
Chicago/Turabian StyleD'Orazio, John, Stuart Jarrett, Alexandra Amaro-Ortiz, and Timothy Scott. 2013. "UV Radiation and the Skin" International Journal of Molecular Sciences 14, no. 6: 12222-12248. https://doi.org/10.3390/ijms140612222
APA StyleD'Orazio, J., Jarrett, S., Amaro-Ortiz, A., & Scott, T. (2013). UV Radiation and the Skin. International Journal of Molecular Sciences, 14(6), 12222-12248. https://doi.org/10.3390/ijms140612222