The Role of AKT/mTOR Pathway in Stress Response to UV-Irradiation: Implication in Skin Carcinogenesis by Regulation of Apoptosis, Autophagy and Senescence

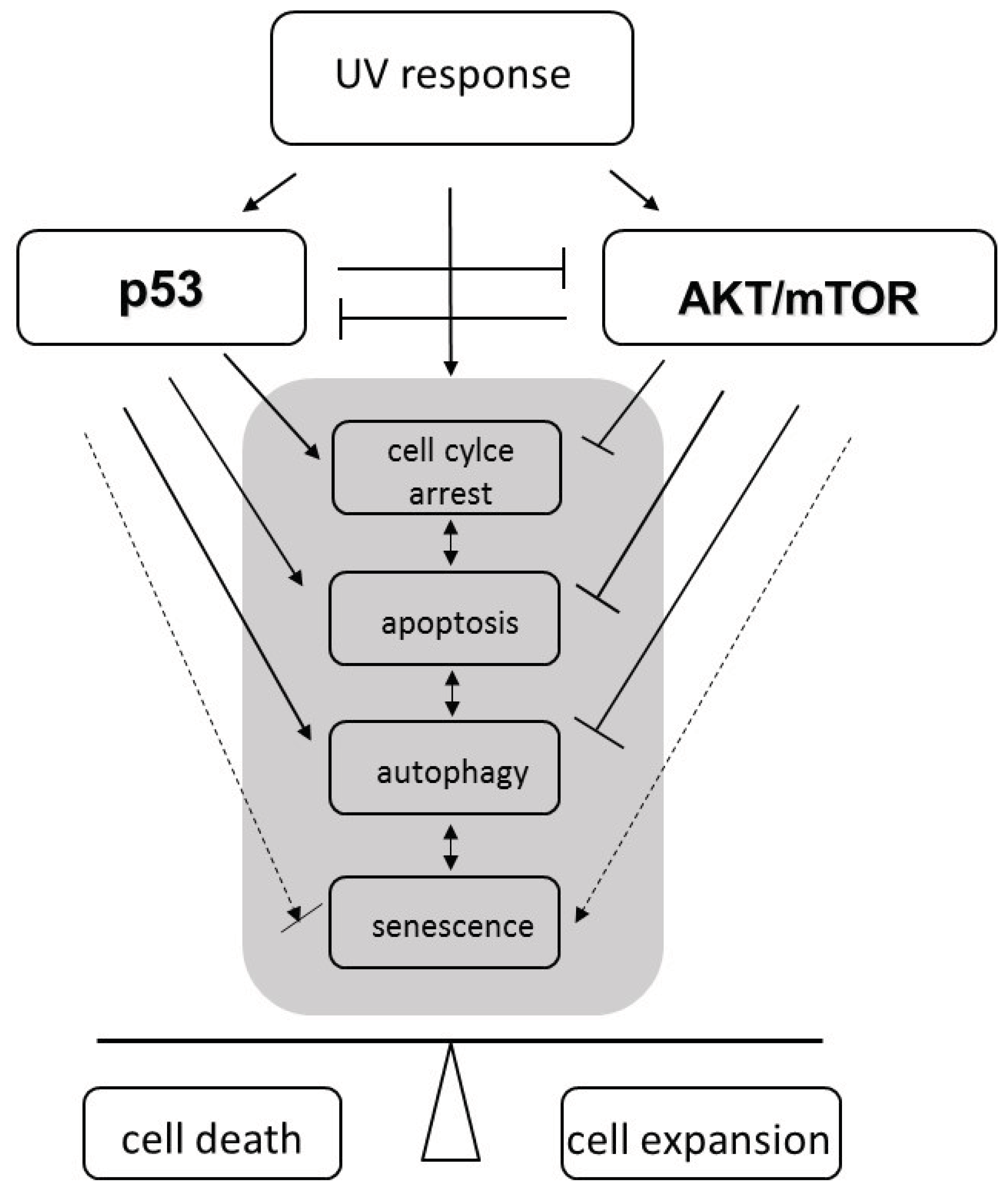{kind=link}
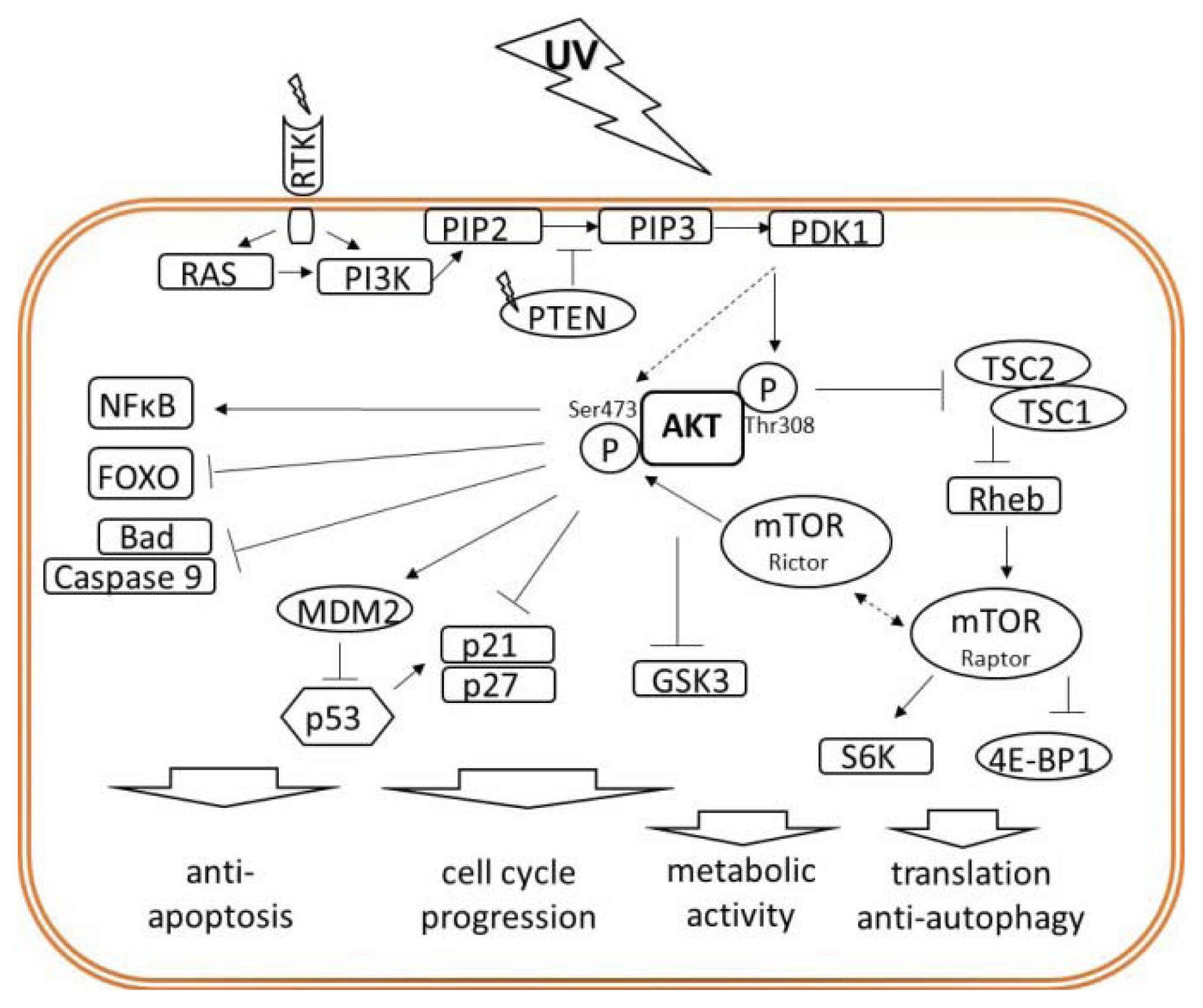{kind=link}
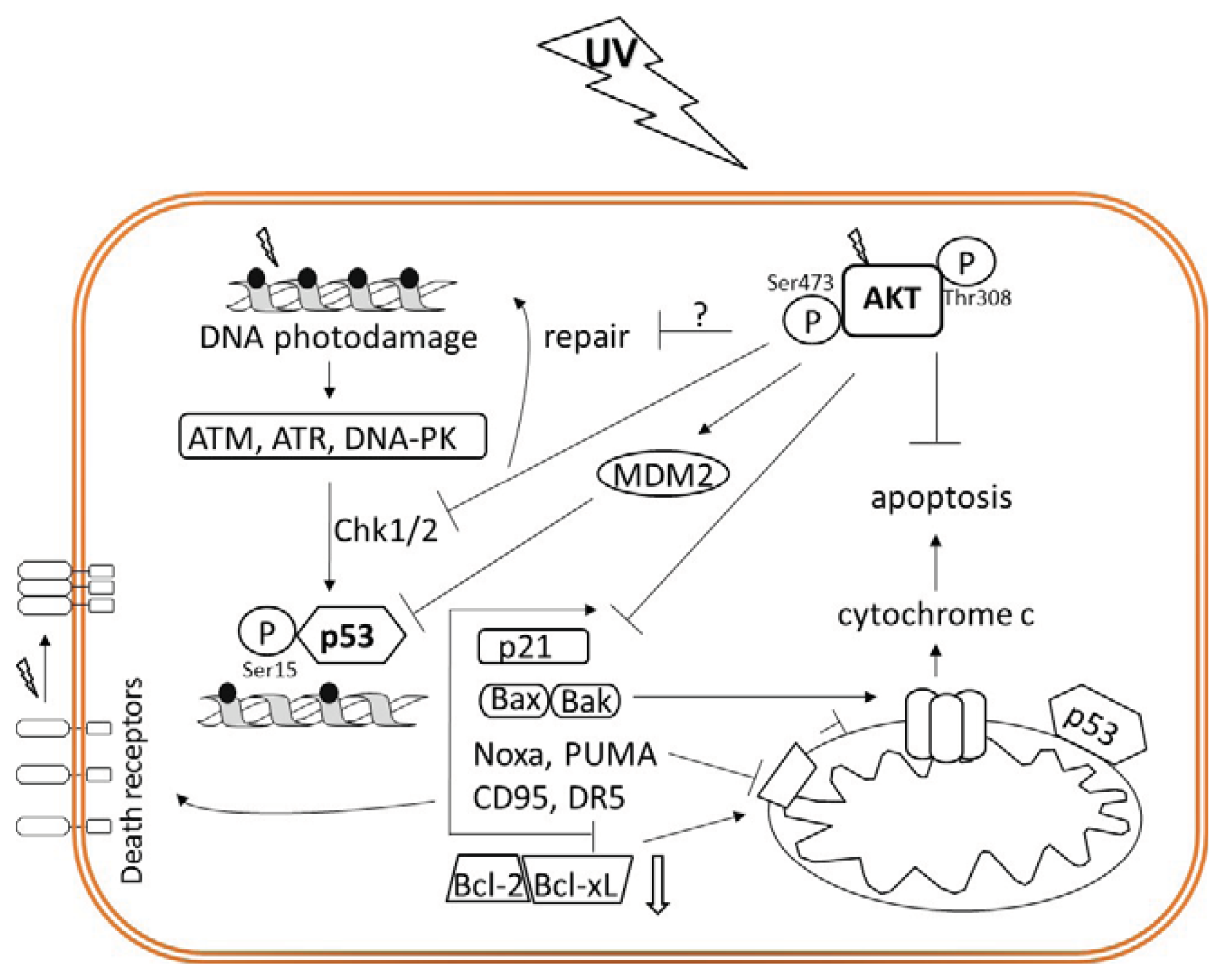{kind=link}
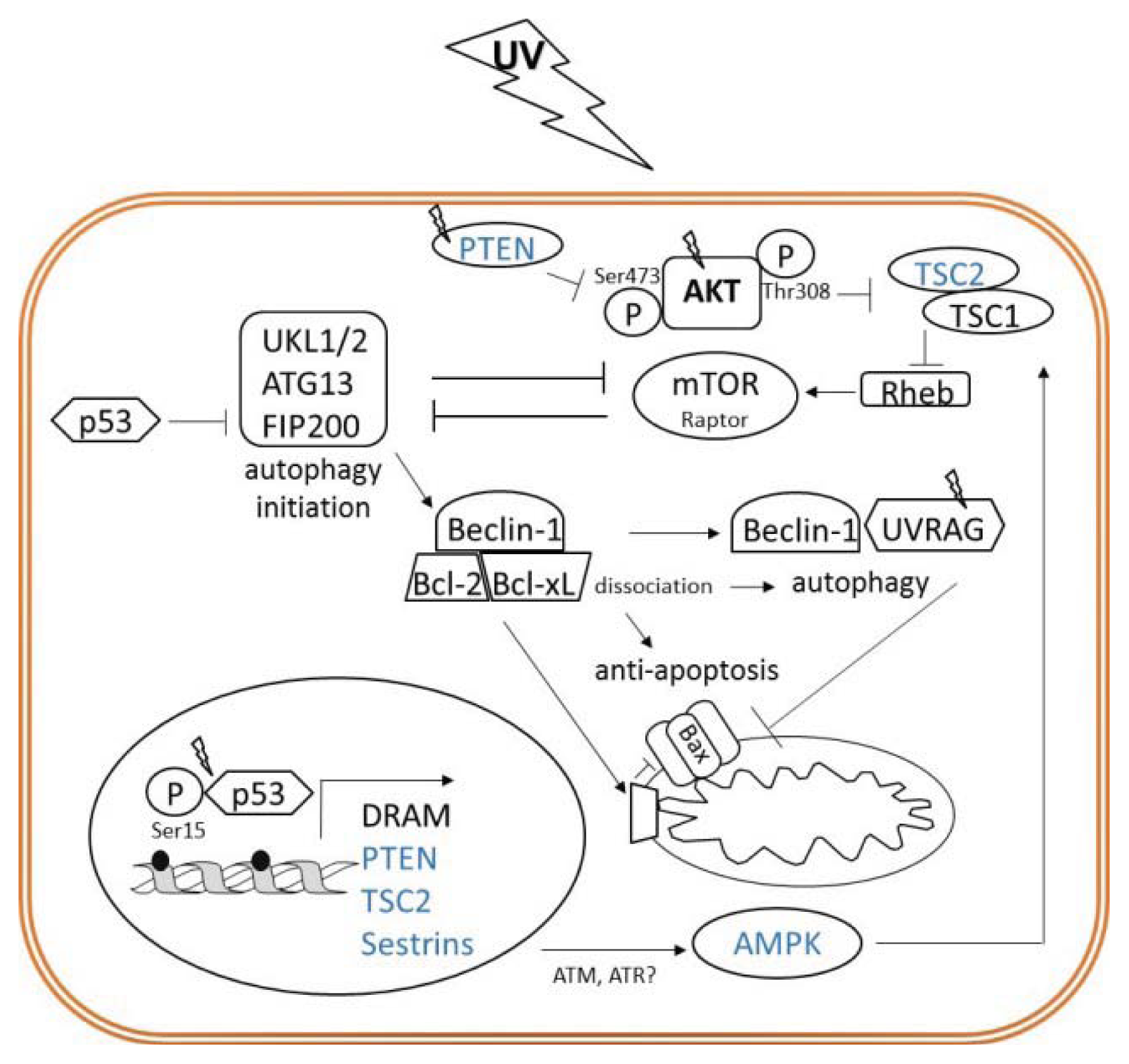{kind=link}
{kind=link}
Abstract
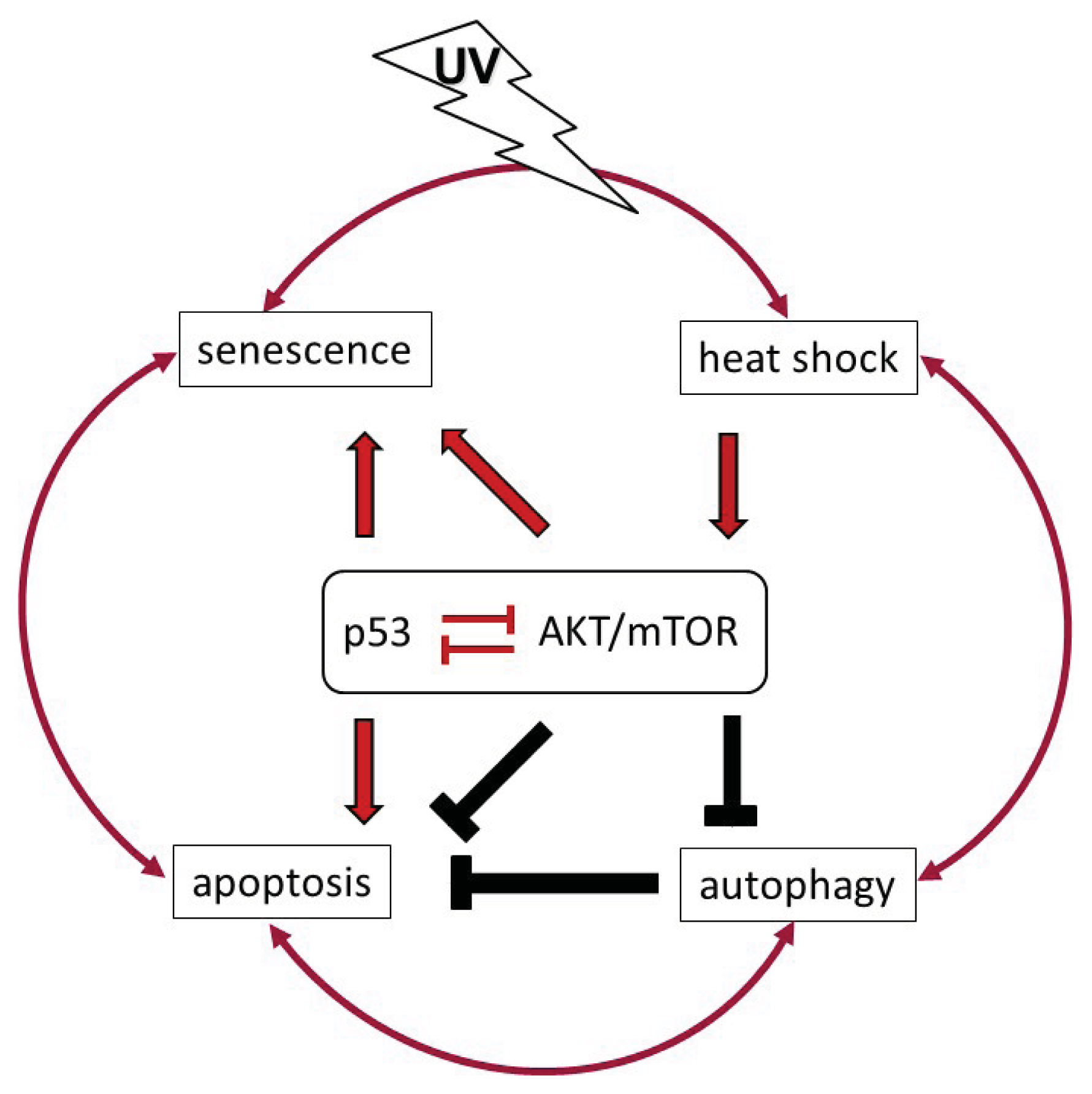
:1. Introduction
2. Mutagenicity of UV Radiation as a Prerequisite for Skin Cancer Development
3. AKT Potently Mediates Oncogenic Signaling
4. Interplay Between AKT and p53 Modulates UV-Induced DNA Damage Responses
5. AKT/mTOR Pathway Impedes UV-Induced Apoptosis
6. Implication of the AKT/mTOR Pathway in Photocarcinogenesis
7. Alternative Roles of p53 and AKT/mTOR Pathways in UV Responses: Autophagy
8. p53-dependent DNA Damage Responses and Oncogenic Pathways Induce Senescence
9. UV Does Not Act Alone: Impact of Heat Shock and Infrared on UV Response
10. Conclusions
Acknowledgments
Conflict of Interest
References
- National Toxicology Program. U.S. Department of Health and Human Services. Public Health Service. Report on carcinogens, 12th edition Available online: http://ntp.niehs.nih.gov/ntp/roc/twelfth/roc12.pdf (accessed on 18 March 2013).
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar]
- Budden, T.; Bowden, N.A. The role of altered nucleotide excision repair and UVB-induced DNA damage in melanomagenesis. Int. J. Mol. Sci 2013, 14, 1132–1151. [Google Scholar]
- Lee, C.-H.; Wu, S.-B.; Hong, C.-H.; Yu, H.-S.; Wei, Y.-H. Molecular mechanisms of UV-induced apoptosis and its effect on skin residential cells: The implication in UV-based phototherapy. Int. J. Mol. Sci 2013, 14, 6414–6435. [Google Scholar]
- Lennikov, A.; Kitaichi, N.; Kase, S.; Noda, K.; Horie, Y.; Nakai, A.; Ohno, S.; Ishida, S. Induction of heat shock protein 70 ameliorates ultraviolet-induced photokeratitis in mice. Int. J. Mol. Sci 2013, 14, 2175–2189. [Google Scholar]
- Nakajima, S.; Lan, S.; Kanno, S.; Takao, M.; Yamamoto, K.; Eker, A.P.; Yasui, A. UV light induced DNA damage and tolerance for the survival of nucleotide excision repair-deficient human cells. J. Biol. Chem 2004, 279, 46674–46677. [Google Scholar]
- Douki, T.; Cadet, J. Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions. Biochemistry 2001, 40, 2495–2501. [Google Scholar]
- Marrot, L.; Belaidi, J.-P.; Meunier, J.-R. Comet assay combined with p53 detection as a sensitive approach for DNA photoprotection assessment in vitro. Exp. Dermatol 2002, 11, 33–36. [Google Scholar]
- De Gruijl, F.R. Photocarcinogenesis: UVA vs. UVB radiation. Skin Pharmacol. Appl. Skin Physiol 2002, 15, 316–320. [Google Scholar]
- Douki, T.; Reynaud-Angelin, A.; Cadet, J.; Sage, E. Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation. Biochemistry 2003, 42, 9221–9226. [Google Scholar]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar]
- Rünger, T.M.; Kappes, U.P. Mechanisms of mutation formation with long-wave ultraviolet light (UVA). Photodermatol. Photoimmunol. Photomed 2008, 24, 2–10. [Google Scholar]
- Besaratinia, T.W.; Synold, H.H.; Chen, C.; Chang, B.; Xi, A.D.; Riggs, G.P.; Pfeifer, G.P. DNA lesions induced by UV A1 and B radiation in human cells: Comparative analyses in the overall genome and in the p53 tumor suppressor gene. Proc. Natl. Acad. Sci. USA 2005, 102, 10058–10063. [Google Scholar]
- Farrell, A.W.; Halliday, G.M.; Lyons, J.G. Chromatin structure following UV-induced DNA damage—Repair or death? Int. J. Mol. Sci 2011, 12, 8063–8085. [Google Scholar]
- Wangari-Talbot, J.; Chen, S. Genetics of melanoma. Front. Genet 2012, 3, 330. [Google Scholar]
- Maddodi, N.; Jayanthy, A.; Setaluri, V. Shining light on skin pigmentation: The darker and the brighter side of effects of UV radiation. Photochem. Photobiol 2012, 88, 1075–1082. [Google Scholar]
- Wang, H.-T.; Choi, B.; Tang, M.-S. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc. Natl. Acad. Sci. USA 2010, 107, 12180–12185. [Google Scholar]
- Noonan, F.P.; Zaidi, M.R.; Wolnicka-Glubisz, A.; Anver, M.R.; Bahn, J.; Wielgus, A.; Cadet, J.; Douki, T.; Mouret, S.; Tucker, M.A.; et al. Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat. Commun 2012, 3, 884. [Google Scholar]
- Jandova, J.; Janda, J.; Sligh, J.E. Changes in mitochondrial DNA alter expression of nuclear encoded genes associated with tumorigenesis. Exp. Cell Res 2012, 318, 2215–2225. [Google Scholar]
- López-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Pérez-Plasencia, C.; Alvarez-Sánchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci 2012, 13, 142–172. [Google Scholar]
- Wan, Y.S.; Wang, Z.Q.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation activates PI 3-kinase/AKT survival pathway via EGF receptors in human skin. Int. J. Oncol 2001, 18, 461–466. [Google Scholar]
- Zhang, Q.S.; Maddock, D.A.; Chen, J.P.; Heo, S.; Chiu, C.; Lai, D.; Souza, K.; Mehta, S.; Wan, Y.S. Cytokine-induced p38 activation feedback regulates the prolonged activation of AKT cell survival pathway initiated by reactive oxygen species in response to UV irradiation in human keratinocytesInt. J. Oncol 2001, 19, 1057–1061. [Google Scholar]
- Iordanov, M.S.; Choi, R.J.; Ryabinina, O.P.; Dinh, T.H.; Bright, R.K.; Magun, B.E. The UV (Ribotoxic) stress response of human keratinocytes involves the unexpected uncoupling of the Ras-extracellular signal-regulated kinase signaling cascade from the activated epidermal growth factor receptor. Mol. Cell Biol 2002, 22, 5380–5394. [Google Scholar]
- Wang, H.Q.; Quan, T.; He, T.; Franke, T.F.; Voorhees, J.J.; Fisher, G.J. Epidermal growth factor receptor-dependent, NF-kappaB-independent activation of the phosphatidylinositol 3-kinase/Akt pathway inhibits ultraviolet irradiation-induced caspases-3, -8, and -9 in human keratinocytes. J. Biol. Chem 2003, 278, 45737–45745. [Google Scholar]
- Han, W.; He, Y.Y. Requirement for metalloproteinase-dependent ERK and AKT activation in UVB-induced G1-S cell cycle progression of human keratinocytes. Photochem. Photobiol 2009, 85, 997–1003. [Google Scholar]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar]
- Xu, N.; Lao, Y.; Zhang, Y.; Gillespie, D.A. Akt: A double-edged sword in cell proliferation and genome stability. J. Oncol 2012, 2012, 951724. [Google Scholar]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar]
- Dijkers, P.F.; Birkenkamp, K.U.; Lam, E.W.; Thomas, N.S.; Lammers, J.W.; Koenderman, L.; Coffer, P.J. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: Protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J. Cell Biol 2002, 156, 531–542. [Google Scholar]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Yamanoshita, O.; Iida, M.; Ohgami, N.; Tamura, H.; Kawamoto, Y.; Kato, M. RAS/RAF/MEK/ERK and PI3K/PTEN/AKT signaling in malignant melanoma progression and therapy. Dermatol. Res. Pract 2012, 2012, 354191. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar]
- Zhang, S.; Yu, D. PI(3)king apart PTEN’s role in cancer. Clin. Cancer Res 2010, 16, 4325–4330. [Google Scholar]
- Jean, C.; Hernandez-Pigeon, H.; Blanc, A.; Charveron, M.; Laurent, G. Epidermal growth factor receptor pathway mitigates UVA-induced G2/M arrest in keratinocyte cells. J. Invest. Dermatol 2007, 127, 2418–2424. [Google Scholar]
- He, Y.Y.; Council, S.E.; Feng, L.; Chignell, C.F. UVA-induced cell cycle progression is mediated by a disintegrin and metalloprotease/epidermal growth factor receptor/AKT/Cyclin D1 pathways in keratinocytes. Cancer Res 2008, 68, 3752–3758. [Google Scholar]
- Madson, J.G.; Lynch, D.T.; Svoboda, J.; Ophardt, R.; Yanagida, J.; Putta, S.K.; Bowles, A.; Trempus, C.S.; Tennant, R.W.; Hansen, L.A. Erbb2 suppresses DNA damage-induced checkpoint activation and UV-induced mouse skin tumorigenesis. Am. J. Pathol 2009, 174, 2357–2366. [Google Scholar]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Chu, W.M.; Guan, K.L.; Wan, Y. AMP-activated protein kinase contributes to UV- and H2O2-induced apoptosis in human skin keratinocytes. J. Biol. Chem 2008, 283, 28897–28908. [Google Scholar]
- Ming, M.; Han, W.; Maddox, J.; Soltani, K.; Shea, C.R.; Freeman, D.M.; He, Y.Y. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene 2010, 29, 492–502. [Google Scholar]
- Zhao, B.; Ming, M.; He, Y.Y. Suppression of PTEN transcription by UVA. J. Biochem. Mol. Toxicol 2013, 27, 184–191. [Google Scholar]
- Gottlieb, T.M.; Martinez Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar]
- Perry, M.E. Mdm2 in the response to radiation. Mol. Cancer Res 2004, 2, 9–19. [Google Scholar]
- Sekulić, A.; Hudson, C.C.; Homme, J.L.; Yin, P.; Otterness, D.M.; Karnitz, L.M.; Abraham, R.T. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 2000, 60, 3504–3513. [Google Scholar]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol 2009, 10, 307–318. [Google Scholar]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol 2011, 13, 1016–1023. [Google Scholar]
- Cao, C.; Wan, Y. Parameters of protection against ultraviolet radiation-induced skin cell damage. J. Cell. Physiol. 2009, 220, 277–284. [Google Scholar]
- Kawasumi, M.; Lemos, B.; Bradner, J.E.; Thibodeau, R.; Kim, Y.S.; Schmidt, M.; Higgins, E.; Koo, S.W.; Angle-Zahn, A.; Chen, A.; et al. Protection from UV-induced skin carcinogenesis by genetic inhibition of the ataxia telangiectasia and Rad3-related (ATR) kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 13716–13721. [Google Scholar]
- Caspari, T. Checkpoints: How to activate p53. Curr. Biol 2000, 10, R315–R317. [Google Scholar]
- Yun, U.J.; Park, H.D.; Shin, D.Y. p53 prevents immature escaping from cell cycle G2 checkpoint arrest through inhibiting cdk2-dependent NF-Y phosphorylation. Cancer Res. Treat 2006, 38, 224–228. [Google Scholar]
- Concin, N.; Stimpfl, M.; Zeillinger, C.; Wolff, U.; Hefler, L.; Sedlak, J.; Leodolter, S.; Zeillinger, R. Role of p53 in G2/M cell cycle arrest and apoptosis in response to gamma-irradiation in ovarian carcinoma cell lines. Int. J. Oncol 2003, 22, 51–57. [Google Scholar]
- Hegde, V.; Yadavilli, S.; Deutsch, W.A. Knockdown of ribosomal protein S3 protects human cells from genotoxic stress. DNA Repair 2007, 6, 94–99. [Google Scholar]
- Fraser, M.; Harding, S.M.; Zhao, H.; Coackley, C.; Durocher, D.; Bristow, R.G. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 2011, 10, 2218–2232. [Google Scholar]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar]
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; Murphy, M.E. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521. [Google Scholar]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem 2001, 276, 7320–7326. [Google Scholar]
- Rass, K.; Reichrath, J. UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer. Adv. Exp. Med. Biol 2008, 624, 162–178. [Google Scholar]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar]
- Tron, V.A.; Trotter, M.J.; Tang, L.; Krajewska, M.; Reed, J.C.; Ho, V.C.; Li, G. p53-regulated apoptosis is differentiation dependent in ultraviolet B-irradiated mouse keratinocytes. Am. J. Pathol 1998, 153, 579–585. [Google Scholar]
- Jonason, A.S.; Kunala, S.; Price, G.J.; Restifo, R.J.; Spinelli, H.M.; Persing, J.A.; Leffell, D.J.; Tarone, R.E.; Brash, D.E. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc. Natl. Acad. Sci. USA 1996, 93, 14025–14029. [Google Scholar]
- Zerp, S.F.; van Elsas, A.; Peltenburg, L.T.; Schrier, P.I. P53 mutations in human cutaneous melanoma correlate with sun exposure but are not always involved in melanomagenesis. Br. J. Cancer 1999, 79, 921–926. [Google Scholar]
- Lehman, T.A.; Modali, R.; Boukamp, P.; Stanek, J.; Bennett, W.P.; Welsh, J.A.; Metcalf, R.A.; Stampfer, M.R.; Fusenig, N.; Rogan, E.M.; et al. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis 1993, 14, 833–839. [Google Scholar]
- Kulms, D.; Zeise, E.; Pöppelmann, B.; Schwarz, T. DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene 2002, 21, 5844–5851. [Google Scholar]
- Paz, M.L.; González Maglio, D.H.; Weill, F.S.; Bustamante, J.; Leoni, J. Mitochondrial dysfunction and cellular stress progression after ultraviolet B irradiation in human keratinocytes. Photodermatol. Photoimmunol. Photomed 2008, 24, 115–122. [Google Scholar]
- Aragane, Y.; Kulms, D.; Metze, D.; Kothny, G.; Pöppelmann, B.; Luger, T.A.; Schwarz, T. Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. J. Cell Biol 1998, 140, 171–182. [Google Scholar]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998, 17, 1675–1687. [Google Scholar]
- Nuñez, G.; Benedict, M.A.; Hu, Y.; Inohara, N. Caspases: The proteases of the apoptotic pathway. Oncogene 1998, 17, 3237–3245. [Google Scholar]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell. Biol 2001, 11, 372–377. [Google Scholar]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar]
- Kothny-Wilkes, G.; Kulms, D.; Luger, T.A.; Kubin, M.; Schwarz, T. Interleukin-1 protects transformed keratinocytes from tumor necrosis factor-related apoptosis-inducing ligand- and CD95-induced apoptosis but not from ultraviolet radiation-induced apoptosis. J. Biol. Chem 1999, 274, 28916–28921. [Google Scholar]
- Pöppelmann, B.; Klimmek, K.; Strozyk, E.; Voss, R.; Schwarz, T.; Kulms, D. NFκB-dependent down-regulation of tumor necrosis factor receptor-associated proteins contributes to interleukin-1-mediated enhancement of ultraviolet B-induced apoptosis. J. Biol. Chem 2005, 280, 15635–15643. [Google Scholar]
- Barisic, S.; Strozyk, E.; Peters, N.; Walczak, H.; Kulms, D. Identification of PP2A as a crucial regulator of the NF-kappaB feedback loop: Its inhibition by UVB turns NF-kappaB into a pro-apoptotic factor. Cell Death Differ. 2008, 15, 1681–1690. [Google Scholar]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell 2011, 145, 92–103. [Google Scholar]
- Strozyk, E.; Pöppelmann, B.; Schwarz, T.; Kulms, D. Differential effects of NF-kappaB on apoptosis induced by DNA-damaging agents: The type of DNA damage determines the final outcome. Oncogene 2006, 25, 6239–6251. [Google Scholar]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch. Dermatol. Res 2010, 302, 5–17. [Google Scholar]
- Ji, C.; Yang, Y.L.; Yang, Z.; Tu, Y.; Cheng, L.; Chen, B.; Xia, J.P.; Sun, W.L.; Su, Z.L.; He, L.; Bi, Z.G. Perifosine sensitizes UVB-induced apoptosis in skin cells: New implication of skin cancer prevention? Cell. Signal 2012, 24, 1781–1789. [Google Scholar]
- Carr, T.D.; DiGiovanni, J.; Lynch, C.J.; Shantz, L.M. Inhibition of mTOR suppresses UVB-induced keratinocyte proliferation and survival. Cancer Prev. Res. (Phila. ) 2012, 5, 1394–1404. [Google Scholar]
- Chen, S.J.; Nakahara, T.; Takahara, M.; Kido, M.; Dugu, L.; Uchi, H.; Takeuchi, S.; Tu, Y.T.; Moroi, Y.; Furue, M. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br. J. Dermatol 2009, 160, 442–445. [Google Scholar]
- Einspahr, J.G.; Calvert, V.; Alberts, D.S.; Curiel-Lewandrowski, C.; Warneke, J.; Krouse, R.; Stratton, S.P.; Liotta, L.; Longo, C.; Pellacani, G.; et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev. Res. (Phila. ) 2012, 5, 403–413. [Google Scholar]
- Kim, J.H.; Baek, S.H.; Kim, D.H.; Choi, T.Y.; Yoon, T.J.; Hwang, J.S.; Kim, M.R.; Kwon, H.J.; Lee, C.H. Downregulation of melanin synthesis by haginin A and its application to in vivo lightening model. J. Invest. Dermatol 2008, 128, 1227–1235. [Google Scholar]
- Syed, D.N.; Afaq, F.; Mukhtar, H. Differential activation of signaling pathways by UVA and UVB radiation in normal humanepidermal keratinocytes. Photochem. Photobiol 2012, 88, 1184–1190. [Google Scholar]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. MTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006, 66, 1500–1508. [Google Scholar]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF- 1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005, 65, 7052–7058. [Google Scholar]
- Liang, C. Negative regulation of autophagy. Cell Death Differ 2010, 17, 1807–1815. [Google Scholar]
- Kroemer, G.; Jäättelä, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar]
- Galluzzi, L.; Vicencio, J.M.; Kepp, O.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. To die or not to die: That is the autophagic question. Curr. Mol. Med 2008, 8, 78–91. [Google Scholar]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol 2010, 12, 823–830. [Google Scholar]
- Aredia, F.; Guamán Ortiz, L.M.; Giansanti, V.; Scovassi, A.I. Autophagy and Cancer. Cells 2012, 1, 520–534. [Google Scholar]
- Chen, N.; Debnath, J. Autophagy and tumorigenesis. FEBS Lett 2010, 584, 1427–1435. [Google Scholar]
- Amelio, I.; Melino, G.; Knight, R.A. Cell death pathology: Cross-talk with autophagy and its clinical implications. Biochem. Biophys. Res. Commun 2011, 414, 277–281. [Google Scholar]
- Chen, L.H.; Chu, P.M.; Lee, Y.J.; Tu, P.H.; Chi, C.W.; Lee, H.C.; Chiou, S.H. Targeting protective autophagy exacerbates UV-triggered apoptotic cell death. Int. J. Mol. Sci 2012, 13, 1209–1224. [Google Scholar]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar]
- Rodriguez-Rocha, H.; Garcia-Garcia, A.; Panayiotidis, M.I.; Franco, R. DNA damage and autophagy. Mutat Res 2011, 711, 158–166. [Google Scholar]
- Meijer, W.H.; van der Klei, I.J.; Veenhuis, M.; Kiel, J.A. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007, 3, 106–116. [Google Scholar]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar]
- Perelman, B.; Dafni, N.; Naiman, T.; Eli, D.; Yaakov, M.; Feng, T.L.; Sinha, S.; Weber, G.; Khodaei, S.; Sancar, A.; et al. Molecular cloning of a novel human gene encoding a 63-kDa protein and its sublocalization within the 11q13 locus. Genomics 1997, 41, 397–405. [Google Scholar]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol 2006, 8, 688–699. [Google Scholar]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000, 19, 5720–5728. [Google Scholar]
- Yang, W.; Ju, J.H.; Lee, K.M.; Nam, K.; Oh, S.; Shin, I. Protein kinase B/Akt1 inhibits autophagy by down-regulating UVRAG expression. Exp. Cell Res 2013, 319, 122–133. [Google Scholar]
- Yin, X.; Cao, L.; Kang, R.; Yang, M.; Wang, Z.; Peng, Y.; Tan, Y.; Liu, L.; Xie, M.; Zhao, Y.; et al. UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep 2011, 12, 727–734. [Google Scholar]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol 2010, 22, 181–185. [Google Scholar]
- Rikiishi, H. Novel insights into the interplay between apoptosis and autophagy. Int. J. Cell Biol 2012, 2012, 317645. [Google Scholar]
- Alexander, A.; Walker, C.L. Differential localization of ATM is correlated with activation of distinct downstream signaling pathways. Cell Cycle 2010, 15, 3685–3686. [Google Scholar]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001, 15, 2177–2196. [Google Scholar]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar]
- Maiuri, M.C.; le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007, 26, 2527–2539. [Google Scholar]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; de Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 2010, 1, e18. [Google Scholar]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar]
- Yang, Y.; Wang, H.; Wang, S.; Xu, M.; Liu, M.; Liao, M.; Frank, J.A.; Adhikari, S.; Bower, K.A.; Shi, X.; et al. GSK3β signaling is involved in ultraviolet B-induced activation of autophagy in epidermal cells. Int. J. Oncol 2012, 41, 1782–1788. [Google Scholar]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar]
- Murase, D.; Hachiya, A.; Takano, K.; Hicks, R.; Visscher, M.O.; Kitahara, T.; Hase, T.; Takema, Y.; Yoshimori, T. Autophagy plays a significant role in determining skin color by regulating melanosome degradation inKeratinocytes. J. Invest. Dermatol. 2013. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 91–101. [Google Scholar]
- Yu, H.; McDaid, R.; Lee, J.; Possik, P.; Li, L.; Kumar, S.M.; Elder, D.E.; van Belle, P.; Gimotty, P.; Guerra, M.; et al. The role of BRAF mutation and p53 inactivation during transformation of a subpopulation of primary human melanocytes. Am. J. Pathol 2009, 174, 2367–2377. [Google Scholar]
- Lewis, D.A.; Yi, Q.; Travers, J.B.; Spandau, D.F. UVB-induced senescence in human keratinocytes requires a functional insulin-like growth factor-1 receptor and p53. Mol. Biol. Cell 2008, 19, 1346–1353. [Google Scholar]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2003. [Google Scholar] [CrossRef]
- Kang, E.S.; Iwata, K.; Ikami, K.; Ham, S.A.; Kim, H.J.; Chang, K.C.; Lee, J.H.; Kim, J.H.; Park, S.B.; Kim, J.H.; et al. Aldose reductase in keratinocytes attenuates cellular apoptosis and senescence induced by UV radiation. Free Radic. Biol. Med 2011, 50, 680–688. [Google Scholar]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 1996, 15, 6541–6551. [Google Scholar]
- Ham, S.A.; Hwang, J.S.; Yoo, T.; Lee, H.; Kang, E.S.; Park, C.; Oh, J.W.; Lee, H.T.; Min, G.; Kim, J.H.; et al. Ligand-activated PPARδ inhibits UVB-induced senescence of human keratinocytes via PTEN-mediated inhibition of superoxide production. Biochem. J 2012, 444, 27–38. [Google Scholar]
- Demidenko, Z.N.; Korotchkina, L.G.; Gudkov, A.V.; Blagosklonny, M.V. Paradoxical suppression of cellular senescence by p53. Proc. Natl. Acad. Sci. USA 2010, 107, 9660–9664. [Google Scholar]
- Dirac, A.M.; Bernards, R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J. Biol. Chem 2003, 278, 11731–11734. [Google Scholar]
- Takahashi, A.; Yamakawa, N.; Mori, E.; Ohnishi, K.; Yokota, S.; Sugo, N.; Aratani, Y.; Koyama, H.; Ohnishi, T. Development of thermotolerance requires interaction between polymerase-beta and heat shock proteins. Cancer Sci 2008, 99, 973–978. [Google Scholar]
- Krawczyk, P.M.; Eppink, B.; Essers, J.; Stap, J.; Rodermond, H.; Odijk, H.; Zelensky, A.; van Bree, C.; Stalpers, L.J.; Buist, M.R.; et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc. Natl. Acad. Sci. USA 2011, 108, 9851–9856. [Google Scholar]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair (Amst. ) 2007, 6, 923–935. [Google Scholar]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol 2010, 2010, 214074. [Google Scholar]
- Li, H.; Liu, L.; Xing, D.; Chen, W.R. Inhibition of the JNK/Bim pathway by Hsp70 prevents Bax activation in UV-induced apoptosis. FEBS Lett 2010, 584, 4672–4678. [Google Scholar]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jäättelä, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol 2001, 3, 839–843. [Google Scholar]
- Gurbuxani, S.; Schmitt, E.; Cande, C.; Parcellier, A.; Hammann, A.; Daugas, E.; Kouranti, I.; Spahr, C.; Pance, A.; Kroemer, G.; et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene 2003, 22, 6669–6678. [Google Scholar]
- Saleh, A.; Srinivasula, S.M.; Balkir, L.; Robbins, P.D.; Alnemri, E.S. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat. Cell Biol 2000, 2, 476–483. [Google Scholar]
- Trautinger, F.; Kokesch, C.; Herbacek, I.; Knobler, R.M.; Kindås-Mügge, I. Overexpression of the small heat shock protein, hsp27, confers resistance to hyperthermia, but not to oxidative stress and UV-induced cell death, in a stably transfected squamous cellcarcinoma cell line. J. Photochem. Photobiol. B 1997, 39, 90–95. [Google Scholar]
- Yoshihisa, Y.; Hassan, M.A.; Furusawa, Y.; Tabuchi, Y.; Kondo, T.; Shimizu, T. Alkannin, HSP70 inducer, protects against UVB-induced apoptosis in human keratinocytes. PLoS One 2012, 7, e47903. [Google Scholar]
- Chang, Z.; Lu, M.; Park, S.M.; Park, H.K.; Kang, H.S.; Pak, Y.; Park, J.S. Functional HSF1 requires aromatic-participant interactions in protecting mouse embryonic fibroblasts against apoptosis via G2 cell cycle arrest. Mol. Cells 2012, 33, 465–470. [Google Scholar]
- Denman, C.J.; McCracken, J.; Hariharan, V.; Klarquist, J.; Oyarbide-Valencia, K.; Guevara-Patiño, J.A.; Le Poole, I.C. HSP70i accelerates depigmentation in a mouse model of autoimmune vitiligo. J. Invest. Dermatol 2008, 128, 2041–2048. [Google Scholar]
- Hoshino, T.; Matsuda, M.; Yamashita, Y.; Takehara, M.; Fukuya, M.; Mineda, K.; Maji, D.; Ihn, H.; Adachi, H.; Sobue, G.; et al. Suppression of melanin production by expression of HSP70. J. Biol. Chem 2010, 285, 13254–13263. [Google Scholar]
- Konishi, H.; Fujiyoshi, T.; Fukui, Y.; Matsuzaki, H.; Yamamoto, T.; Ono, Y.; Andjelkovic, M.; Hemmings, B.A.; Kikkawa, U. Activation of protein kinase B induced by H2O2 and heat shock through distinct mechanisms dependent and independent of phosphatidylinositol 3-kinase. J. Biochem 1999, 126, 1136–1143. [Google Scholar]
- Matsuzaki, H.; Yamamoto, T.; Kikkawa, U. Distinct activation mechanisms of protein kinase B by growth-factor stimulation and heat-shock treatment. Biochemistry 2004, 43, 4284–4293. [Google Scholar]
- Mustafi, S.B.; Chakraborty, P.K.; Dey, R.S.; Raha, S. Heat stress upregulates chaperone heat shock protein 70 and antioxidant manganase superoxide dismutase through reactive oxygene species (ROS), p38MAPK, and Akt. Cell Stress Chaperones 2009, 14, 579–589. [Google Scholar]
- Jantschitsch, C.; Majewski, S.; Maeda, A.; Schwarz, T.; Schwarz, A. Infrared radiation confers resistance to UV-induced apoptosis via reduction of DNA damage and upregulation of antiapoptotic proteins. J. Invest. Dermatol 2009, 129, 1271–1279. [Google Scholar]
- Jantschitsch, C.; Weichenthal, M.; Maeda, A.; Proksch, E.; Schwarz, T.; Schwarz, A. Infrared radiation does not enhance the frequency of ultraviolet radiation-induced skin tumors, but their growth behaviour in mice. Exp. Dermatol 2011, 20, 346–350. [Google Scholar]





© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Strozyk, E.; Kulms, D. The Role of AKT/mTOR Pathway in Stress Response to UV-Irradiation: Implication in Skin Carcinogenesis by Regulation of Apoptosis, Autophagy and Senescence. Int. J. Mol. Sci. 2013, 14, 15260-15285. https://doi.org/10.3390/ijms140815260
Strozyk E, Kulms D. The Role of AKT/mTOR Pathway in Stress Response to UV-Irradiation: Implication in Skin Carcinogenesis by Regulation of Apoptosis, Autophagy and Senescence. International Journal of Molecular Sciences. 2013; 14(8):15260-15285. https://doi.org/10.3390/ijms140815260
Chicago/Turabian StyleStrozyk, Elwira, and Dagmar Kulms. 2013. "The Role of AKT/mTOR Pathway in Stress Response to UV-Irradiation: Implication in Skin Carcinogenesis by Regulation of Apoptosis, Autophagy and Senescence" International Journal of Molecular Sciences 14, no. 8: 15260-15285. https://doi.org/10.3390/ijms140815260
APA StyleStrozyk, E., & Kulms, D. (2013). The Role of AKT/mTOR Pathway in Stress Response to UV-Irradiation: Implication in Skin Carcinogenesis by Regulation of Apoptosis, Autophagy and Senescence. International Journal of Molecular Sciences, 14(8), 15260-15285. https://doi.org/10.3390/ijms140815260