Green Fluorescent Protein (GFP)-Based Overexpression Screening and Characterization of AgrC, a Receptor Protein of Quorum Sensing in Staphylococcus aureus


Abstract
:1. Introduction
2. Results and Discussion
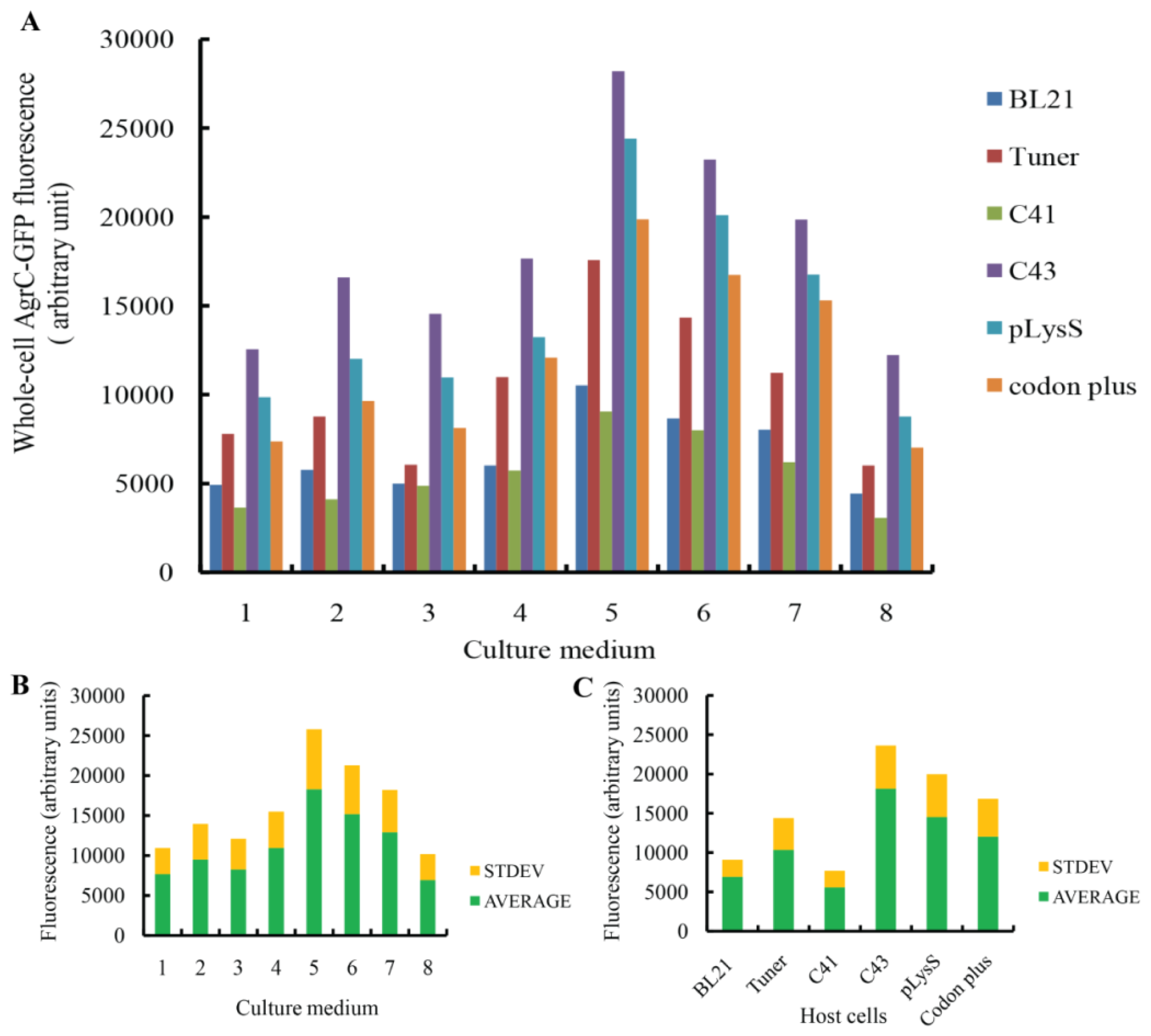
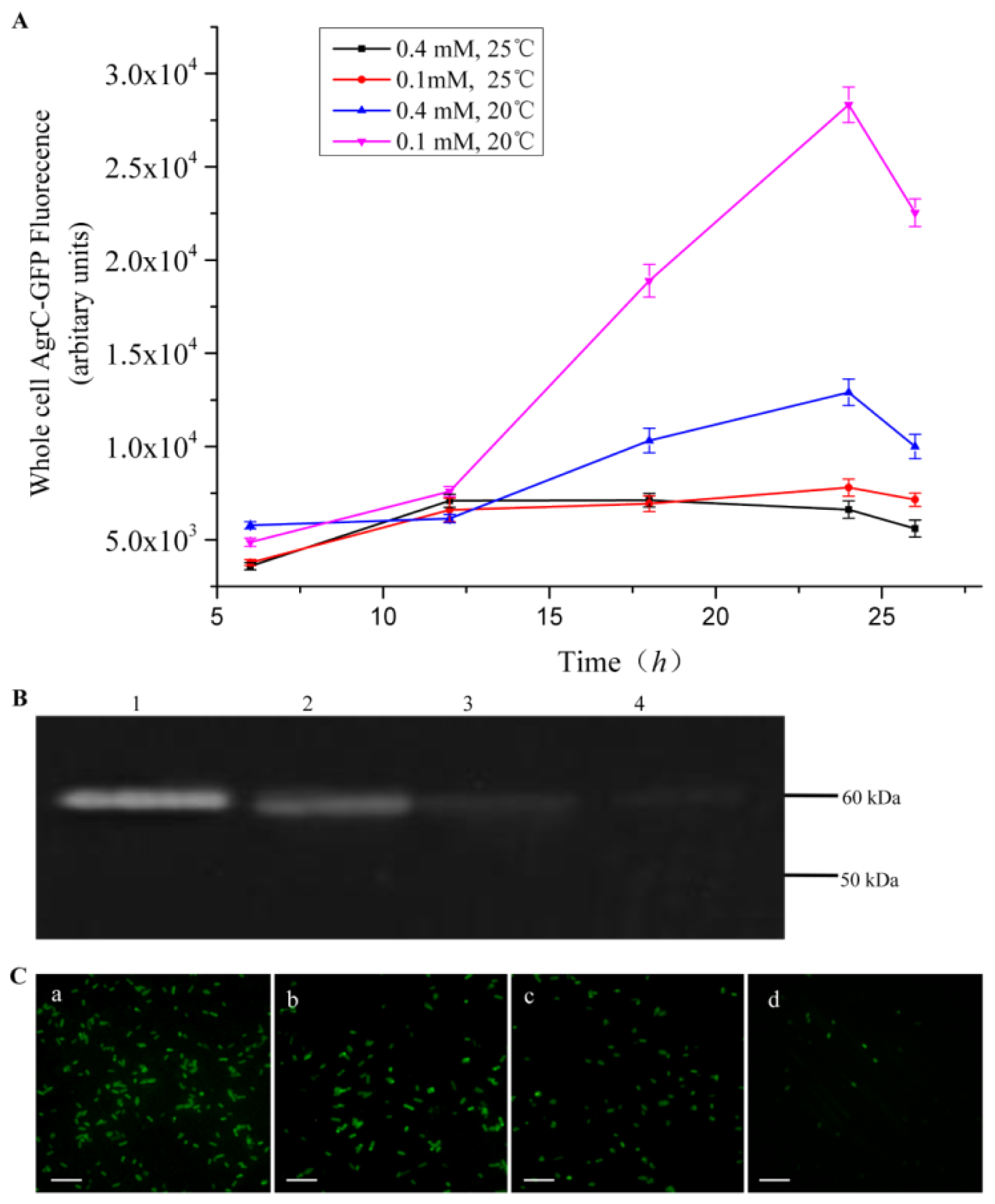
2.1. Expression Screening and Optimization
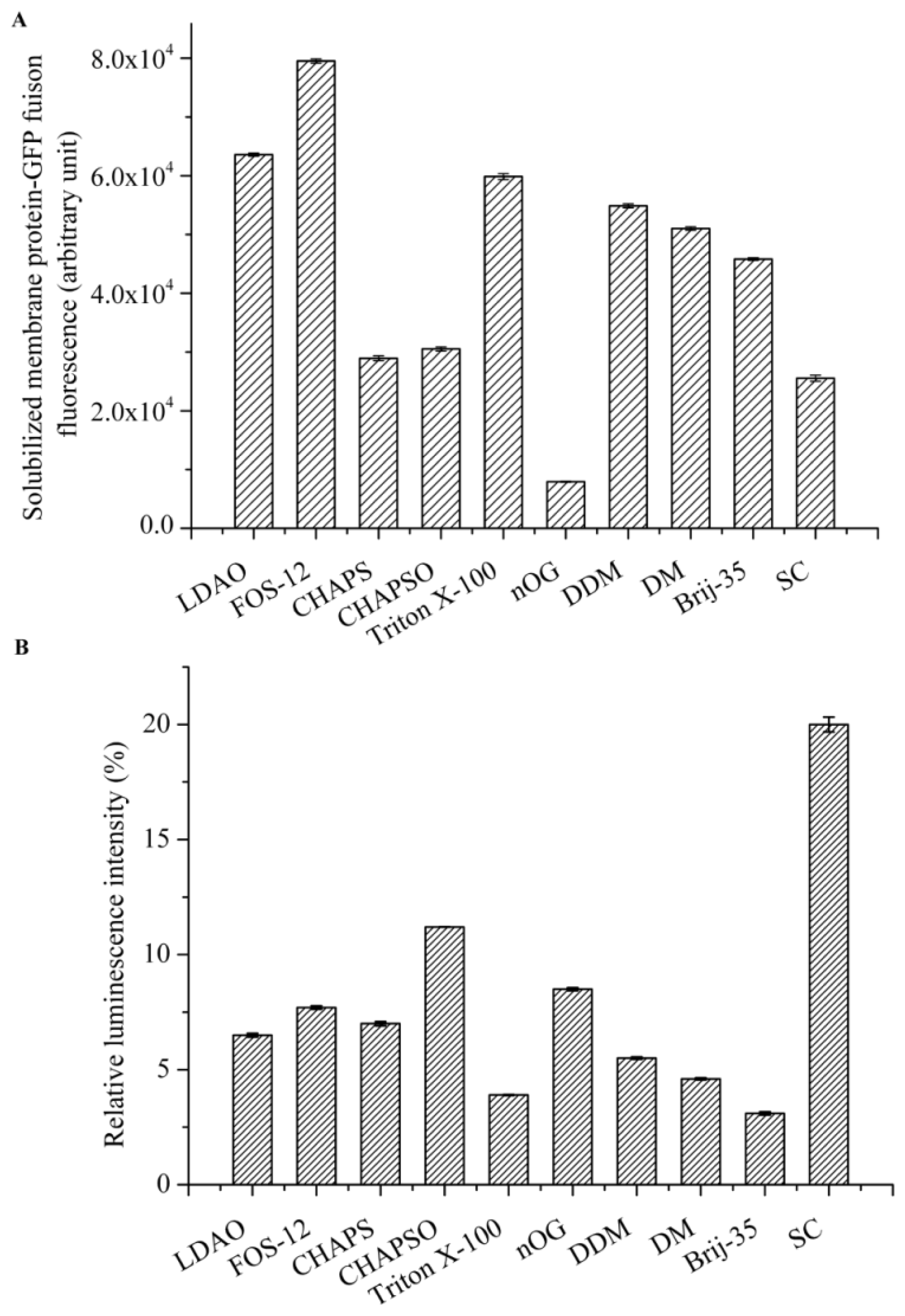
2.2. Detergent Evaluation Based on GFP Fluorescence and Kinase Activity
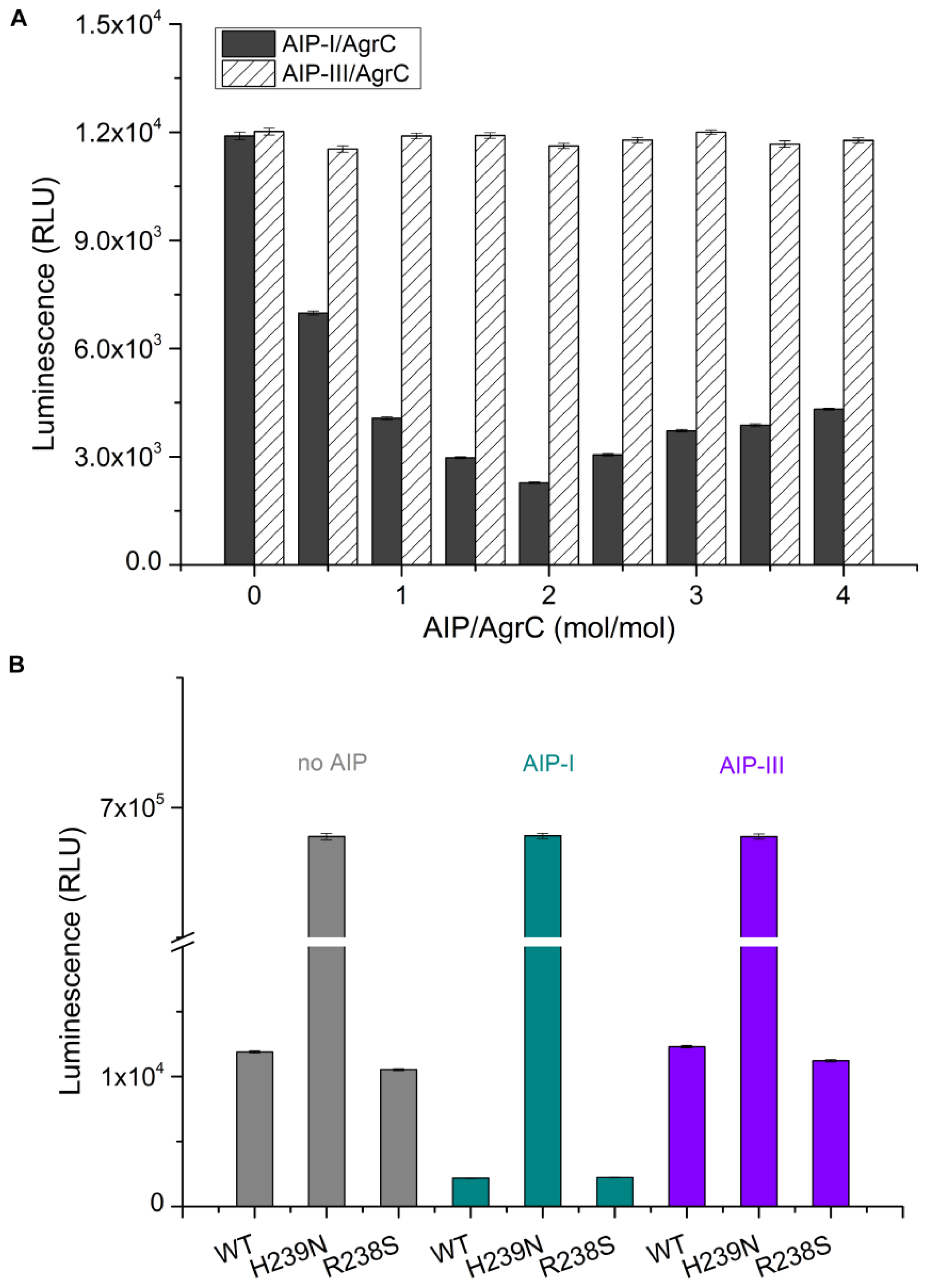
2.3. Stimulation of Autophosphorylztion in Purified AgrC by the Signalling Molecule AIP
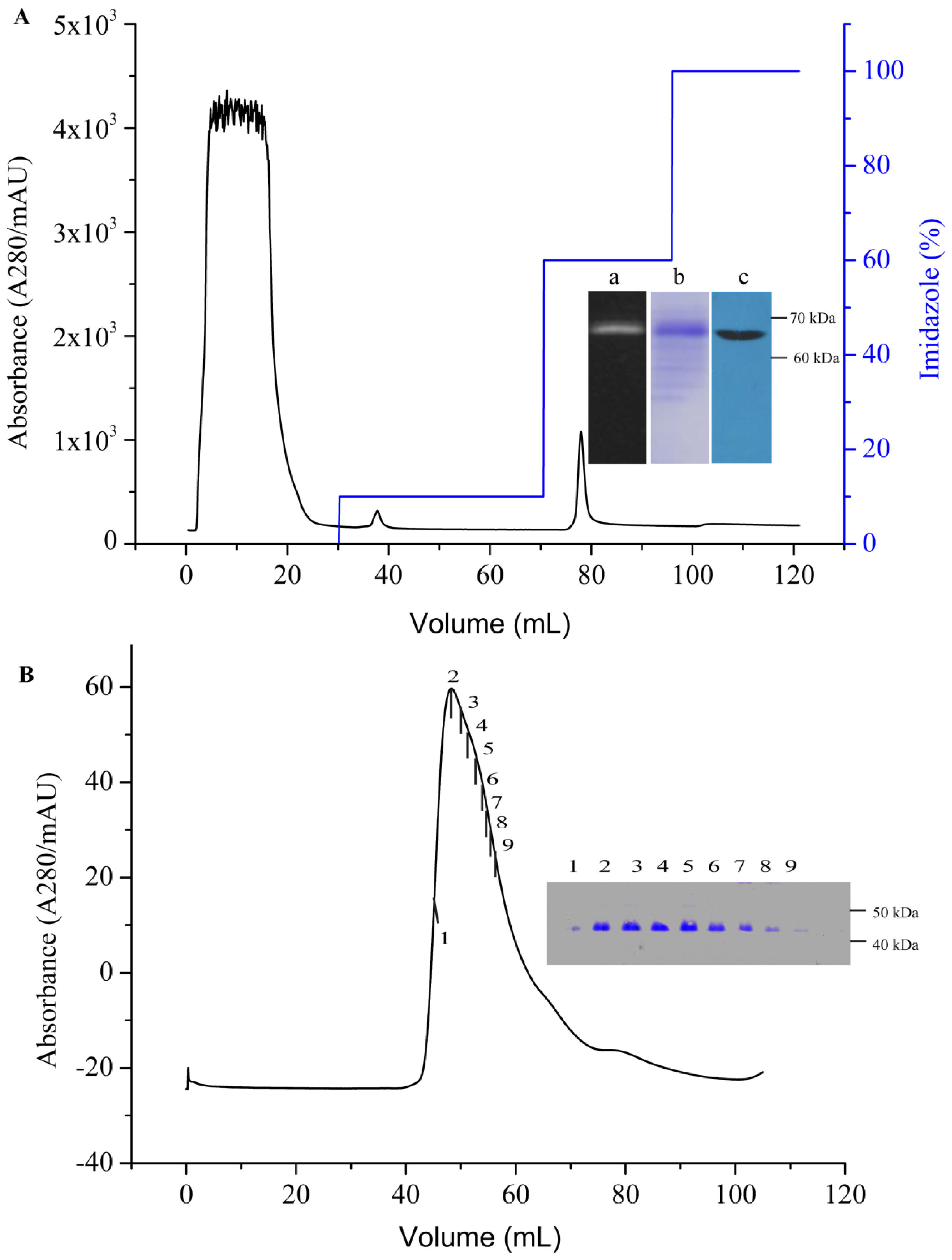
2.4. Purification of AgrC-GFP Based on IMAC and SEC
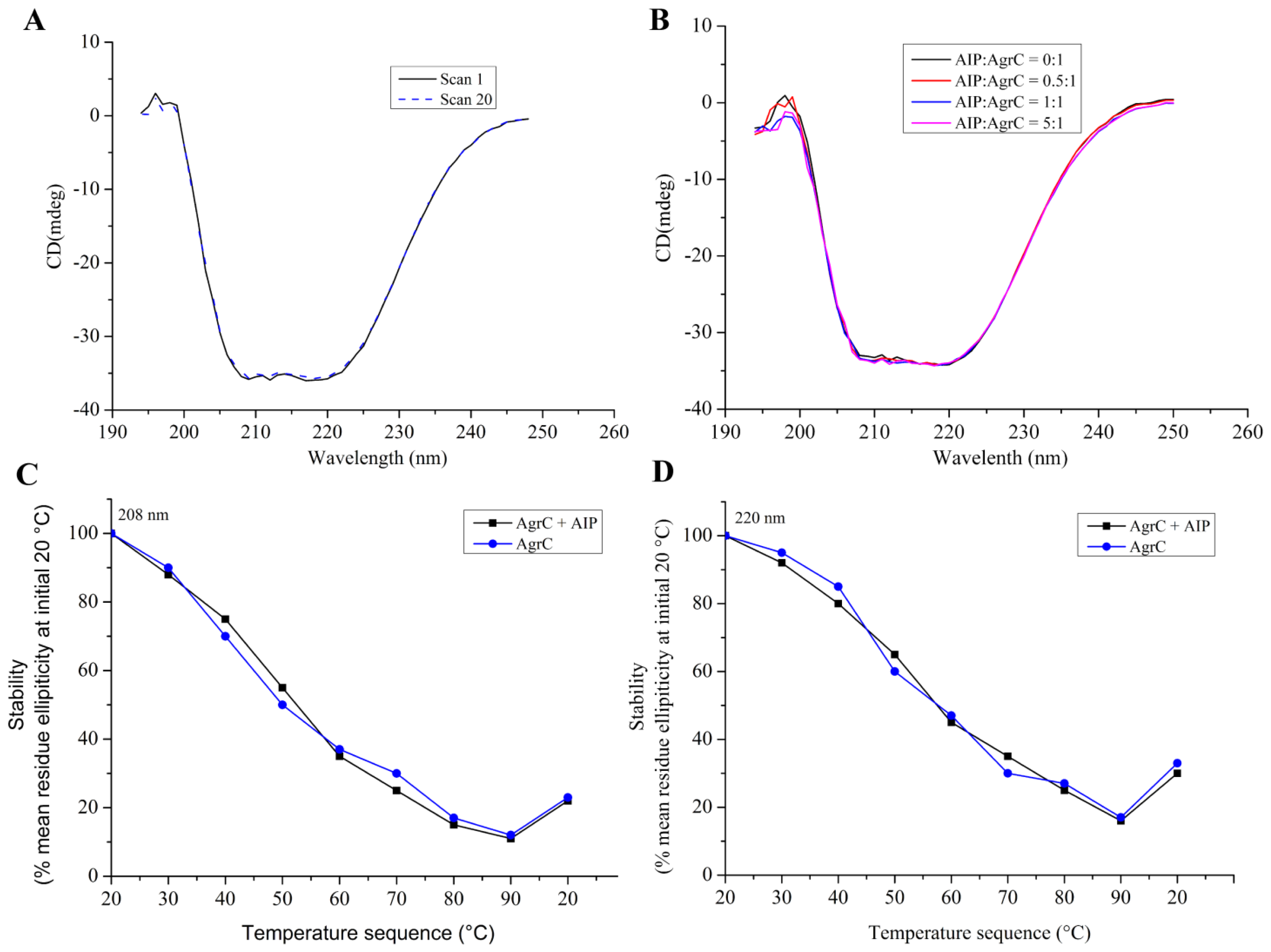
2.5. Secondary Structure and Structural Stabilization of Purified AgrC
3. Experimental Section
3.1. Plasmid Construction
3.2. Expression Screening of AgrC–GFP
3.3. Fluorescence Measurement
3.4. Cell Disruption
3.5. SDS-PAGE and Western Blotting Analysis
3.6. Detergent Selection
3.7. Purification of AgrC–GFP
3.8. Circular Dichroism (CD) Spectroscopy
4. Conclusions
Supplementary Information
ijms-14-18470-s001.pdfAcknowledgments
Conflicts of Interest
References
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol 2001, 55, 165–199. [Google Scholar]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol 2005, 21, 319–346. [Google Scholar]
- Novick, R.P.; Geisinger, E. Quorum sensing in Staphylococci. Annu. Rev. Genet 2008, 42, 541–564. [Google Scholar]
- Pazy, Y.; Motaleb, M.A.; Guarnieri, M.T. Identical phosphatase mechanisms achieved through distinct modes of binding phosphoprotein substrate. Proc. Natl. Acad. Sci. USA 2010, 107, 1924–1929. [Google Scholar]
- Laub, M.T.; Goulian, M. Specificity in two-component signal transduction pathways. Annu. Rev. Genet 2007, 41, 121–145. [Google Scholar]
- Calva, E.; Oropeza, R. Two-component signal transduction systems, environmental signals, and virulence. Microb. Ecol 2006, 51, 166–176. [Google Scholar]
- Bahn, Y.S. Master and commander in fungal pathogens: The two-component system and the HOG signaling pathway. Eukaryot. Cell 2008, 7, 2017–2036. [Google Scholar]
- Thoendel, M.; Kavanaugh, J.S.; Flack, C.E. Peptide signaling in the staphylococci. Chem. Rev 2011, 111, 117–151. [Google Scholar]
- Geisinger, E.; George, E.A.; Muir, T.W.; Novick, R.P. Identification of ligand specificity determinants in AgrC, the Staphylococcus aureus quorum-sensing receptor. J. Biol. Chem 2008, 283, 8930–8938. [Google Scholar]
- Lyon, G.J.; Wright, J.S.; Muir, T.W.; Novick, R.P. Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus. Biochemistry 2002, 41, 10095–10104. [Google Scholar]
- Chen, L.C.; Tsou, L.T.; Chen, F.J. Ligand-receptor recognition for activation of quorum sensing in Staphylococcus aureus. J. Microbiol 2009, 47, 572–581. [Google Scholar]
- Jensen, R.O.; Winzer, K.; Clarke, S.R. Differential recognition of Staphylococcus aureus quorum-sensing signals depends on both extracellular loops 1 and 2 of the transmembrane sensor AgrC. J. Mol. Biol 2008, 381, 300–309. [Google Scholar]
- Geisinger, E.; Muir, T.W.; Novick, R.P. Agr receptor mutants reveal distinct modes of inhibition by staphylococcal autoinducing peptides. Proc. Natl. Acad. Sci. USA 2009, 106, 1216–1221. [Google Scholar]
- Drew, D.E.; von Heijne, G.; Nordlund, P. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett 2001, 507, 220–224. [Google Scholar]
- Drew, D.; Slotboom, D.J.; Friso, G. A scalable, GFP-based pipeline for membrane protein overexpression screening and purification. Protein Sci 2005, 14, 2011–2017. [Google Scholar]
- Drew, D.; Lerch, M.; Kunji, E. Optimization of membrane protein overexpression and purification using GFP fusions. Nat. Methods 2006, 3, 303–313. [Google Scholar]
- Waldo, G.S.; Standish, B.M.; Berendzen, J. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol 1999, 17, 691–695. [Google Scholar]
- Geertsma, E.R.; Groeneveld, M.D.; Slotboom, J. Quality control of overexpressed membrane proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 5722–5727. [Google Scholar]
- Drew, D.; Newstead, S.; Sonoda, Y. GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat. Protoc 2008, 3, 784–798. [Google Scholar]
- Wagner, S.; Klepsch, M.M.; Schlegel, S. Tuning Escherichia coli for membrane protein overexpression. Proc. Natl. Acad. Sci. USA 2008, 105, 14371–14376. [Google Scholar]
- Gordon, E.; Horsefield, R.; Swarts, H.G. Effective high-throughput overproduction of membrane proteins in Escherichia coli. Protein Expr. Purif 2008, 62, 1–8. [Google Scholar]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif 2005, 41, 207–234. [Google Scholar]
- Privé, G.G. Detergents for the stabilization and crystallization. Methods 2007, 41, 388–397. [Google Scholar]
- Fan, J.; Heng, J.; Dai, S.; Shaw, N. An efficient strategy for high throughput screening of recombinant integral membrane protein expression and stability. Protein Expr. Purif 2011, 78, 6–13. [Google Scholar]
- Gräslund, S.; Nordlund, P.; Weigelt, J. Protein production and purification. Nat. Methods 2008, 5, 135–146. [Google Scholar]
- Lundback, A.K.; van den Berg, S.; Hebert, H.; Berglund, H.; Eshaghi, S. Exploring the activity of tobacco etch virus protease in detergent solutions. Anal. Biochem 2008, 382, 69–71. [Google Scholar]
- Mohanty, A.K.; Simmons, C.R.; Wiener, M.C. Inhibition of tobacco etch virus protease activity by detergents. Protein Expr. Purif 2003, 27, 109–114. [Google Scholar]
- Hammon, J.; Palanivelu, D.V.; Chen, J.; Patel, C.; Minor, D.L., Jr. A green fluorescent protein screen for identification of well-expressed membrane proteins from a cohort of extremophilic organisms. Protein Sci. 2009, 18, 121–133. [Google Scholar]
- Chen, C.P.; Kernytsky, A.; Rost, B. Transmembrane helix predictions revisited. Protein Sci 2002, 11, 2774–2791. [Google Scholar]
- Daley, D.O.; Rapp, M.; Granseth, E. Global topology analysis of the Escherichia coli inner membrane proteome. Science 2005, 308, 1321–1323. [Google Scholar]
- Bernsel, A.; Viklund, H.; Hennerdal, A.; Elofsson, A. TOPCONS: Consensus prediction of membrane protein topology. Nucleic Acids Res 2009, 37, W465–W468. [Google Scholar]
- Wiener, M.C. A pedestrian guide to membrane protein crystallization. Methods 2004, 34, 364–372. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Medium name | Medium component |
|---|---|---|
| 1 | LBE | 0.5% (w/v) yeast extract, 1% (w/v) peptone, 0.5% (w/v) glucose, 1% (w/v) NaCl, 2 mM MgSO4, 20 mM KH2PO4, 80 mM K2HPO4 |
| 2 | LBE-LP | 0.5% (w/v) yeast extract, 1% (w/v) peptone, 0.1% (w/v) glucose, 0.5% (w/v) NaCl, 2 mM MgSO4, 5 mM KH2PO4, 20 mM K2HPO4 |
| 3 | LBE-MP | 0.5% (w/v) yeast extract, 1% (w/v) peptone, 0.5% (w/v) glucose, 0.07% (w/v) Na2SO4, 0.25% (w/v) NH4Cl, 2 mM MgSO4, 10 mM KH2PO4, 40 mM K2HPO4 |
| 4 | 2× TYE | 1% (w/v) yeast extract, 1.6% (w/v) peptone, 0.5% (w/v) glucose, 0.5% (w/v) NaCl, 2 mM MgSO4, 20 mM KH2PO4, 80 mM K2HPO4 |
| 5 | TBE | 2.4 (w/v) yeast extract, 1.2% (w/v) peptone, 0.4% (v/v) glycerol, 2 mM MgSO4, 20 mM KH2PO4, 80 mM K2HPO4 |
| 6 | 32Y | 3.2% (w/v) yeast extract, 0.8% (w/v) peptone and 0.58% (w/v) NaCl in 10 mM Tris–HCl pH 7.6 |
| 7 | TB | 2.4% (w/v) yeast extract, 1.2% (w/v) peptone, 0.4% (v/v) glycerol, 20 mM KH2PO4, 80 mM K2HPO4 |
| 8 | 4× TY | 2% (w/v) yeast extract, 0.8% (w/v) peptone and 0.58% (w/v) NaCl |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, L.; Quan, C.; Liu, B.; Xu, Y.; Zhao, P.; Xiong, W.; Fan, S. Green Fluorescent Protein (GFP)-Based Overexpression Screening and Characterization of AgrC, a Receptor Protein of Quorum Sensing in Staphylococcus aureus. Int. J. Mol. Sci. 2013, 14, 18470-18487. https://doi.org/10.3390/ijms140918470
Wang L, Quan C, Liu B, Xu Y, Zhao P, Xiong W, Fan S. Green Fluorescent Protein (GFP)-Based Overexpression Screening and Characterization of AgrC, a Receptor Protein of Quorum Sensing in Staphylococcus aureus. International Journal of Molecular Sciences. 2013; 14(9):18470-18487. https://doi.org/10.3390/ijms140918470
Chicago/Turabian StyleWang, Lina, Chunshan Quan, Baoquan Liu, Yongbin Xu, Pengchao Zhao, Wen Xiong, and Shengdi Fan. 2013. "Green Fluorescent Protein (GFP)-Based Overexpression Screening and Characterization of AgrC, a Receptor Protein of Quorum Sensing in Staphylococcus aureus" International Journal of Molecular Sciences 14, no. 9: 18470-18487. https://doi.org/10.3390/ijms140918470
APA StyleWang, L., Quan, C., Liu, B., Xu, Y., Zhao, P., Xiong, W., & Fan, S. (2013). Green Fluorescent Protein (GFP)-Based Overexpression Screening and Characterization of AgrC, a Receptor Protein of Quorum Sensing in Staphylococcus aureus. International Journal of Molecular Sciences, 14(9), 18470-18487. https://doi.org/10.3390/ijms140918470