3D QSAR Studies, Pharmacophore Modeling and Virtual Screening on a Series of Steroidal Aromatase Inhibitors

Abstract
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | General Structure | Substituents | IC50 (µM) | pIC50 |
|---|---|---|---|---|
| 1 b |  |  | 5.2 | 5.284 |
| 2 a |  | 22.7 | 4.644 | |
| 3 |  | 6.4 | 5.194 | |
| 4 |  | –CH3 | 5.2 | 5.284 |
| 5 a | –CH2CH3 | 18.1 | 4.742 | |
| 6 | –CH2C≡CCH3 | 0.1123 | 6.950 | |
| 7 b | –CH2C≡CCH2CH3 | 0.0118 | 7.928 | |
| 8 a | –CH2C≡C(CH2)2CH3 | 0.083 | 7.081 | |
| 9 | –CH2C≡C(CH2)3CH3 | 0.1811 | 6.742 | |
| 10 | –CH2C≡C(CH2)6CH3 | 2.18 | 5.662 | |
| 11 | –CH2C≡CCH2OH | 0.02 | 7.699 | |
| 12 b (Exemestane) |  | 0.0501 | 7.300 | |
| 13 b (Formestane) |  | 0.0486 | 7.313 | |
| 14 a |  | 3.30 | 5.481 | |
| 15 |  | 0.18 | 6.745 | |
| 16 b |  | 0.16 | 6.796 | |
| 17 |  | 4.90 | 5.310 | |
| 18 a |  | 0.183 | 6.738 | |
| 19 b |  | 0.135 | 6.870 | |
| 20 |  | 0.970 | 6.013 | |
| 21 |  | 0.225 | 6.648 | |
| 22 a |  | 0.145 | 6.839 | |
| 23 |  | 1.733 | 5.761 | |
| 24 |  | 1.150 | 5.939 | |
| 25 a |  | 0.59 | 6.229 | |
| 26 |  | 0.75 | 6.125 | |
| 27 b |  | 0.45 | 6.347 | |
| 28 |  | 0.47 | 6.328 | |
| 29 a |  | 1-OH; 2-Ph | 7.27 | 5.138 |
| 30 | 1-OH; 2-MeOPh(p) | 7.13 | 5.147 | |
| 31 | 1-OCOCF3; 2-Ph | 7.12 | 5.148 | |
| 32 | 1-OCOCF3; 2-MeOPh(p) | 7.00 | 5.155 | |
| 33 |  | 1-OCOCF3; 2-COCH3; 3-Ph | 6.57 | 5.182 |
| 34 a | 1-OCOCF3; 2-COCH3; 3-MeOPh(p) | 6.45 | 5.190 | |
| 35 | 1-OCOCF3; 2-COCH2CH3; 3-Ph | 6.72 | 5.173 | |
| 36 | 1-OCOCF3; 2-COCH2CH3; 3-MeOPh(p) | 6.61 | 5.180 | |
| 37 | 1-OH; 2-H; 3-Ph | 6.91 | 5.161 | |
| 38 | 1-OH; 2-H; 3-MeOPh(p) | 6.83 | 5.166 | |
| 39 a | 1-OCOCF3; 2-CH3; 3-Ph | 5.81 | 5.236 | |
| 40 | 1-OCOCF3; 2-CH3; 3-MeOPh(p) | 5.78 | 5.238 | |
| 41 a | 1-OCOCF3; 2-Ph; 3-Ph | 5.67 | 5.246 | |
| 42 | 1-OCOCF3; 2-Ph; 3-MeOPh(p) | 5.45 | 5.264 | |
| 43 | 1-OH; 2-CH3; 3-Ph | 6.34 | 5.198 | |
| 44 | 1-OH; 2-CH3; 3-MeOPh(p) | 6.12 | 5.213 | |
| 45 | 1-OH; 2-Ph; 3-Ph | 6.01 | 5.221 | |
| 46 a | 1-OH; 2-Ph; 3-MeOPh(p) | 5.92 | 5.228 | |
| 47 |  | 1-H; 2-Ph | 5.23 | 5.281 |
| 48 | 1-H; 2-MeOPh(p) | 4.88 | 5.312 | |
| 49 | 1-COCH3; 2-Ph | 4.91 | 5.309 | |
| 50 | 1-COCH3; 2-MeOPh(p) | 4.89 | 5.311 | |
| 51 a | 1-CH3; 2-Ph | 4.78 | 5.321 | |
| 52 | 1-CH3; 2-MeOPh(p) | 4.56 | 5.341 | |
| 53 | 1-Ph; 2-Ph | 4.27 | 5.370 | |
| 54 | 1-Ph; 2-MeOPh(p) | 4.16 | 5.381 | |
| 55 a |  | 1-COCH3; 2-Ph | 2.88 | 5.541 |
| 56 | 1-COCH3; 2-MeOPh(p) | 2.65 | 5.577 | |
| 57 | 1-H; 2-Ph | 3.01 | 5.521 | |
| 58 | 1-H; 2-MeOPh(p) | 2.91 | 5.536 | |
| 59 | 1-CH3; 2-Ph | 2.45 | 5.611 | |
| 60 a | 1-CH3; 2-MeOPh(p) | 2.11 | 5.676 | |
| 61 | 1-Ph; 2-Ph | 1.98 | 5.703 | |
| 62 b | 1-Ph; 2-MeOPh(p) | 1.82 | 5.740 | |
| 63 |  | 1-CH3; 2-Ph | 3.51 | 5.455 |
| 64 | 1-CH3; 2-MeOPh(p) | 3.40 | 5.469 | |
| 65 a | 1-Ph; 2-Ph | 3.34 | 5.476 | |
| 66 | 1-Ph; 2-MeOPh(p) | 3.23 | 5.491 |
2. Results and Discussion
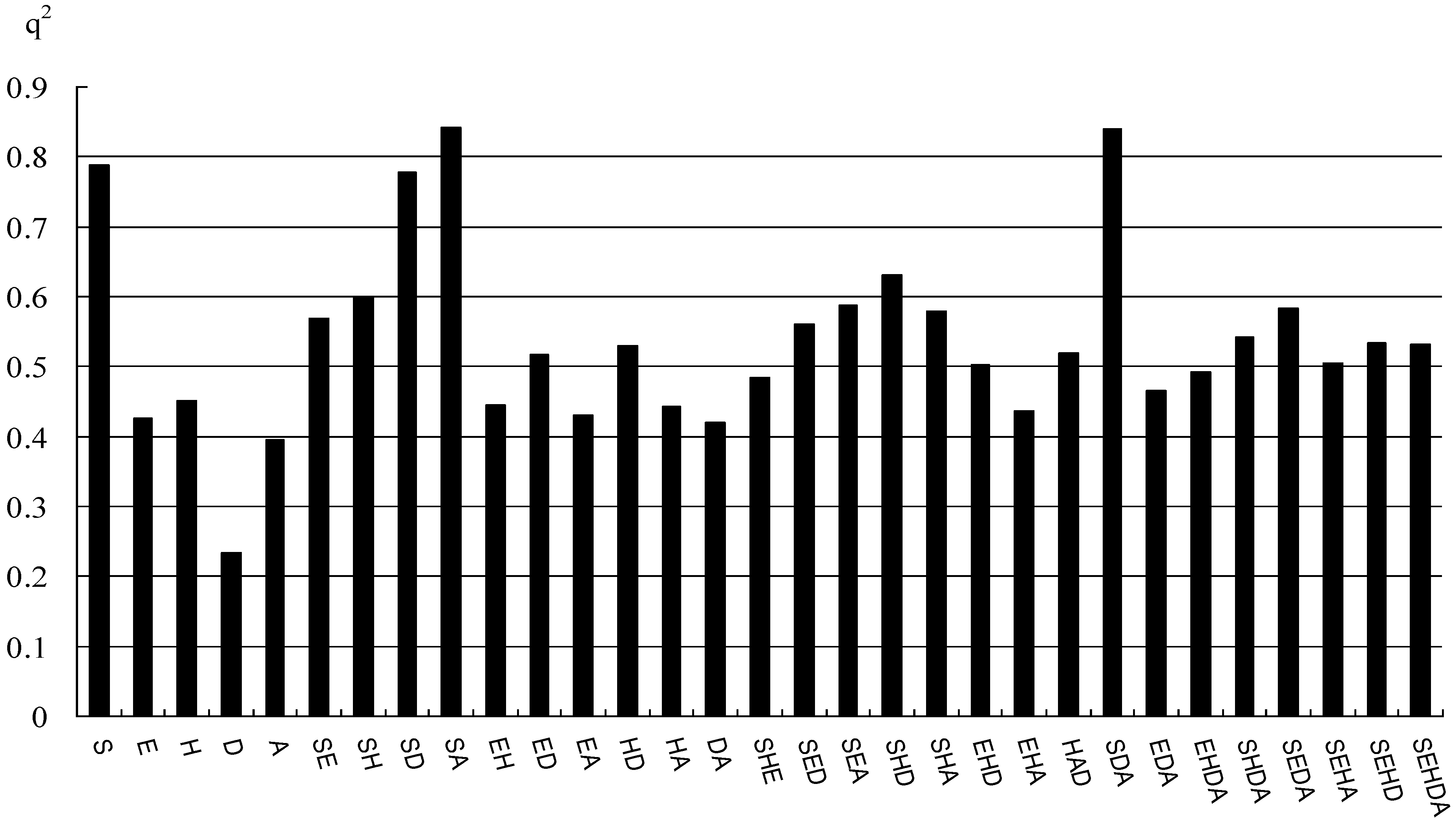
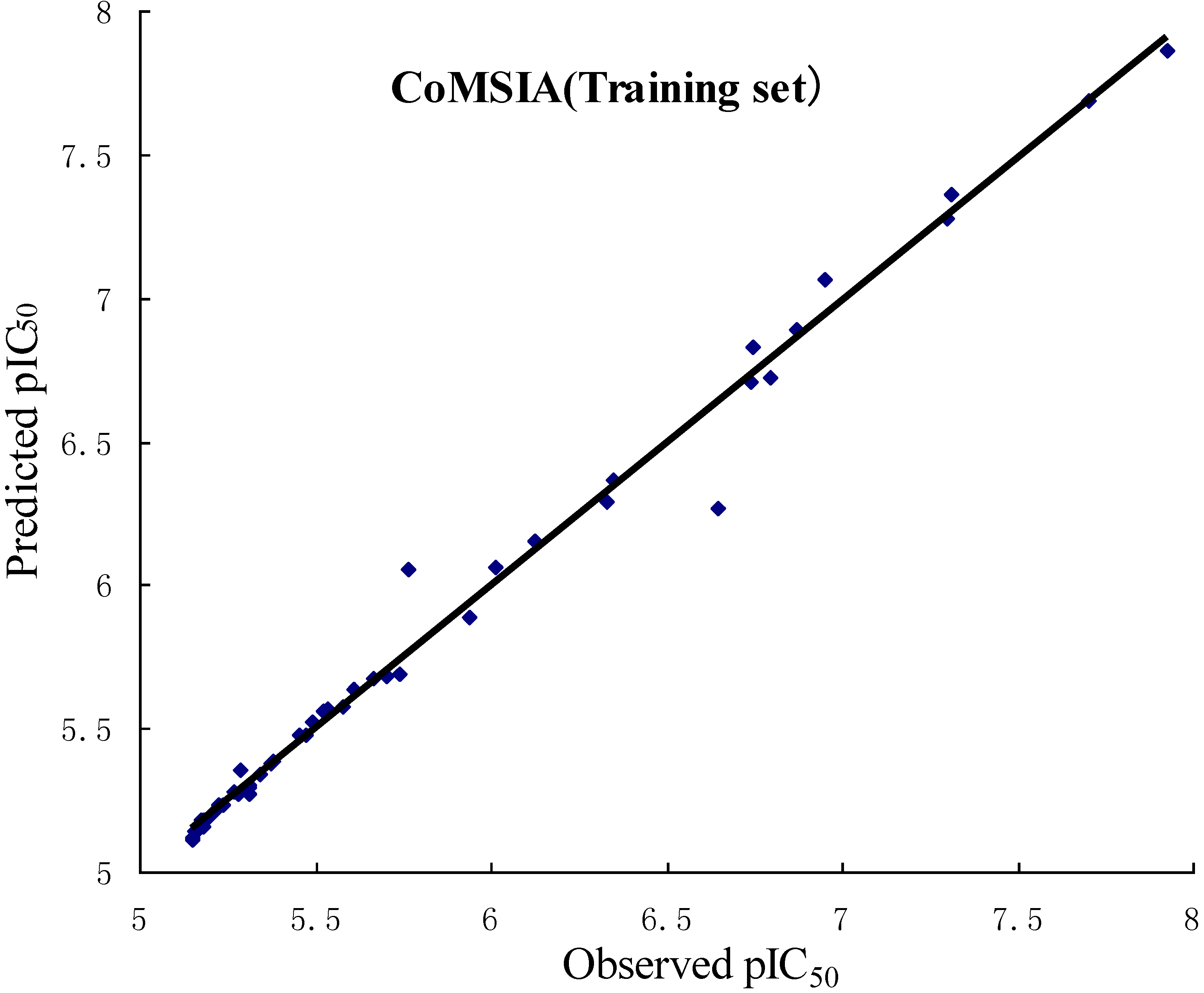
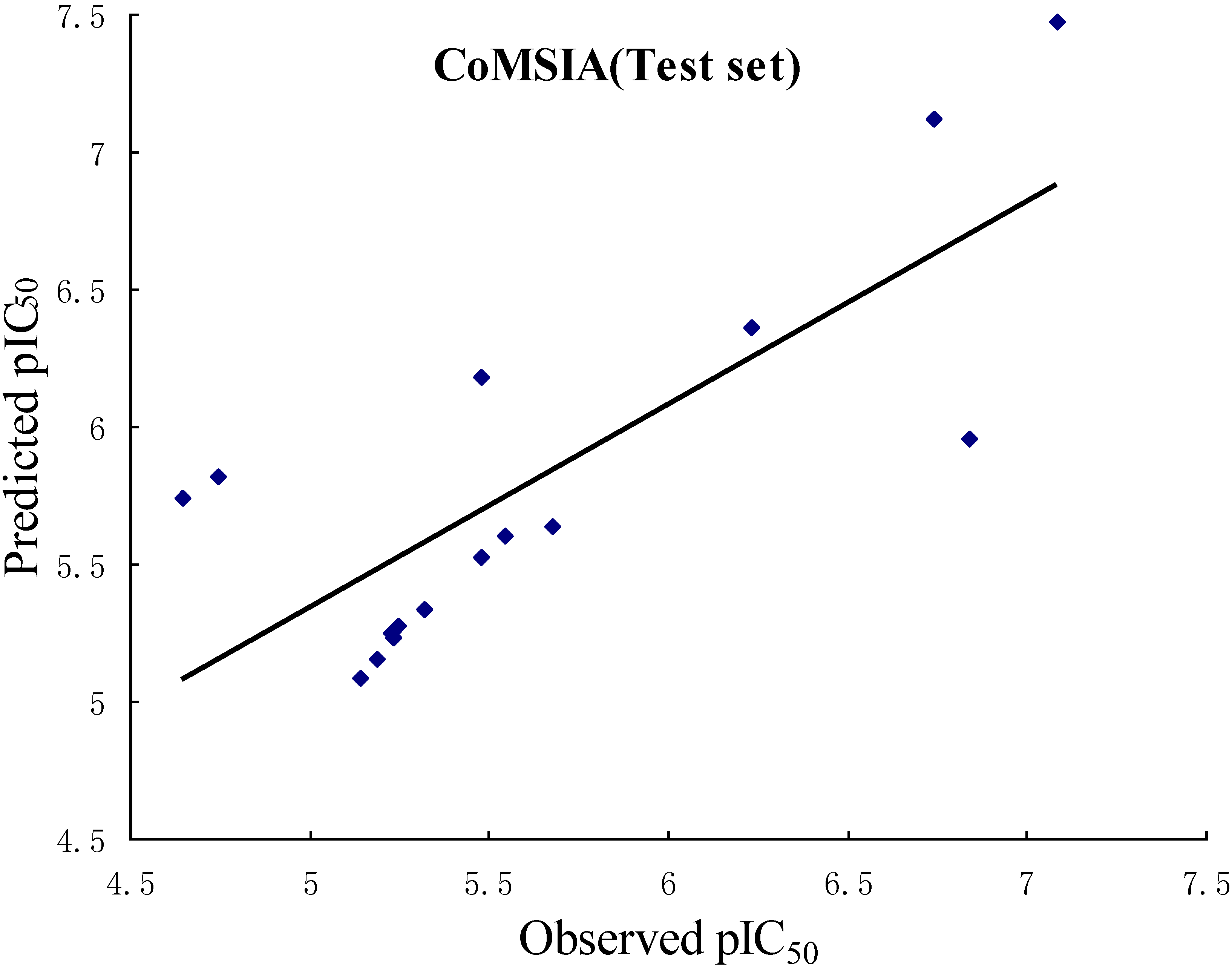
2.1. CoMFA and CoMSIA Statistical Results
| Components | CoMFA | CoMSIA (SA) |
|---|---|---|
| q2 (r2cv) | 0.636 | 0.843 |
| r2ncv | 0.988 | 0.989 |
| SEE | 0.094 | 0.096 |
| F value | 309.026 | 174.304 |
| r2pred | 0.658 | 0.601 |
| No. of compounds | 50 | 50 |
| No. of optimal components | 10 | 17 |
| Contribution | ||
| Steric | 0.671 | 0.677 |
| Electrostatic | 0.329 | – |
| Hydrophobic | – | – |
| H-bond donor | – | – |
| H-bond acceptor | – | 0.323 |

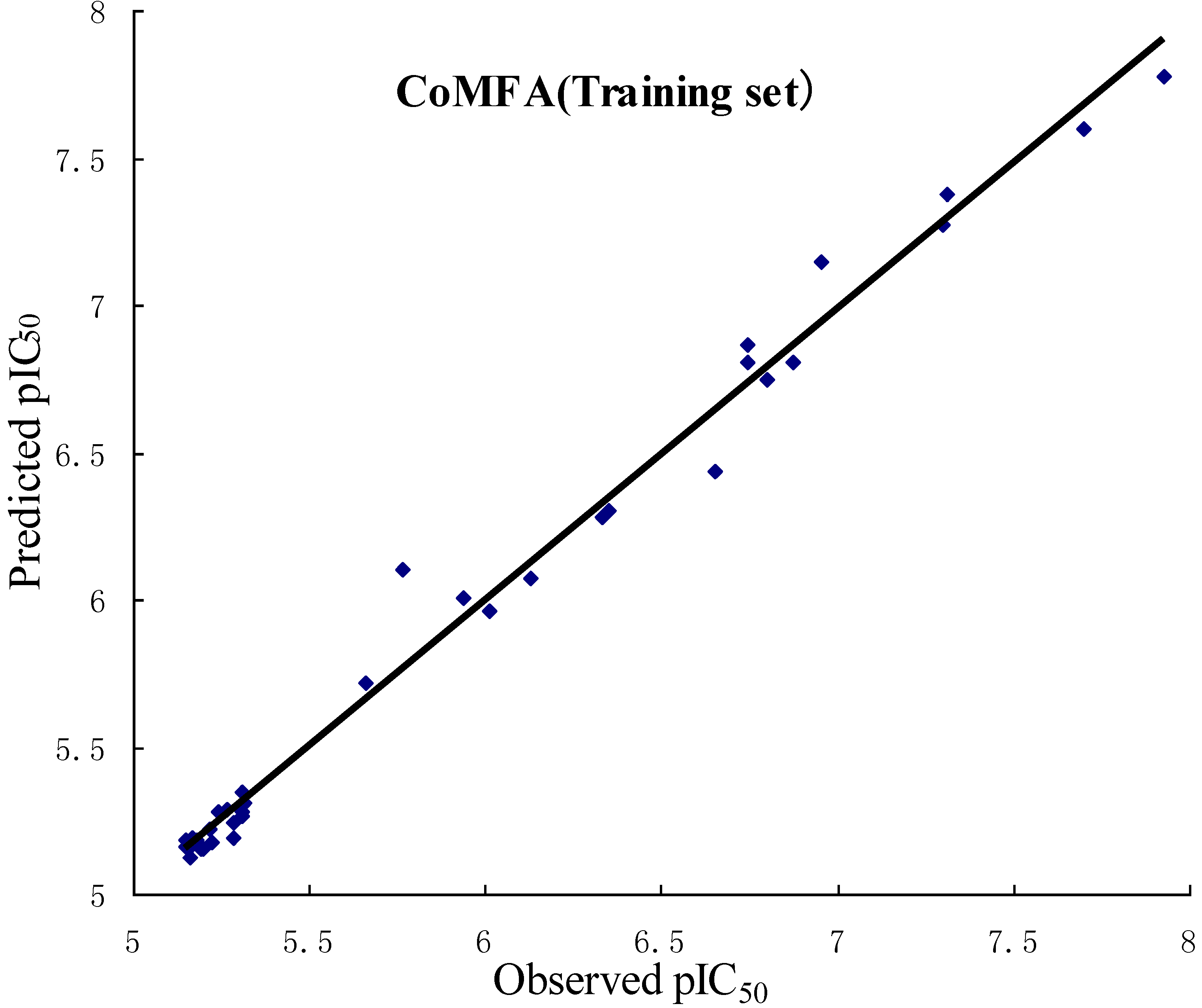
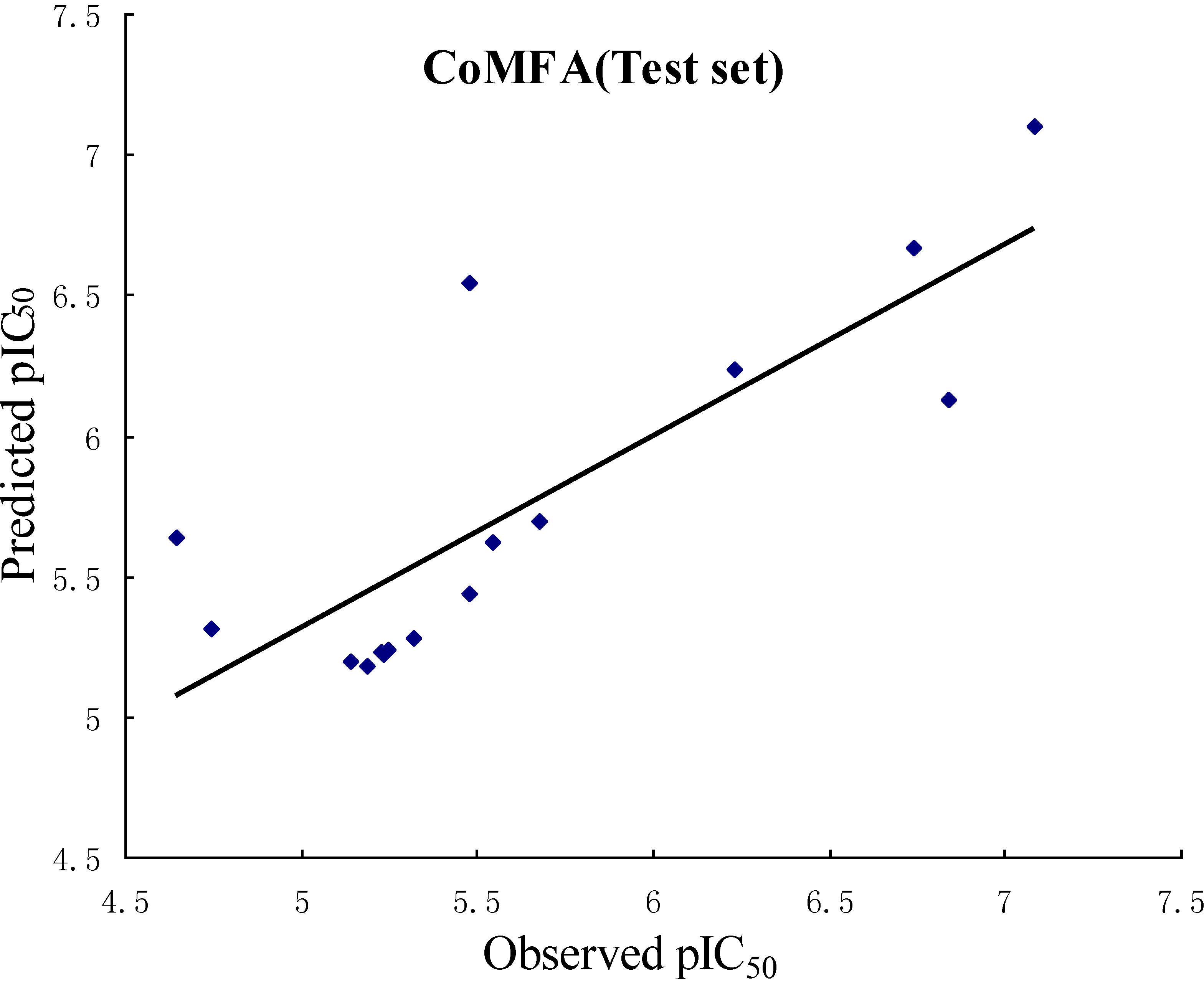
2.2. Validation of 3D QSAR Models
| Compound | Observed pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| Pred. | Res. | Pred. | Res. | ||
| 1 | 5.284 | 5.247 | 0.037 | 5.357 | −0.073 |
| 2 a | 4.644 | 5.642 | −0.998 | 5.744 | −1.100 |
| 3 | 5.194 | 5.152 | 0.042 | 5.192 | 0.002 |
| 4 | 5.284 | 5.191 | 0.093 | 5.278 | 0.006 |
| 5 a | 4.742 | 5.314 | −0.572 | 5.822 | −1.080 |
| 6 | 6.950 | 7.149 | −0.199 | 7.064 | −0.114 |
| 7 | 7.928 | 7.778 | 0.150 | 7.867 | 0.061 |
| 8 a | 7.081 | 7.102 | −0.021 | 7.477 | −0.396 |
| 9 | 6.742 | 6.804 | −0.062 | 6.711 | 0.031 |
| 10 | 5.662 | 5.722 | −0.060 | 5.678 | −0.016 |
| 11 | 7.699 | 7.598 | 0.101 | 7.685 | 0.014 |
| 12 | 7.300 | 7.272 | 0.028 | 7.281 | 0.019 |
| 13 | 7.313 | 7.378 | −0.065 | 7.361 | −0.048 |
| 14 a | 5.481 | 6.542 | −1.061 | 6.181 | −0.700 |
| 15 | 6.745 | 6.869 | −0.124 | 6.830 | −0.085 |
| 16 | 6.796 | 6.746 | 0.050 | 6.726 | 0.070 |
| 17 | 5.310 | 5.348 | −0.038 | 5.307 | 0.003 |
| 18 a | 6.738 | 6.668 | 0.070 | 7.123 | −0.385 |
| 19 | 6.870 | 6.811 | 0.059 | 6.888 | −0.018 |
| 20 | 6.013 | 5.965 | 0.048 | 6.062 | −0.049 |
| 21 | 6.648 | 6.436 | 0.212 | 6.267 | 0.381 |
| 22 a | 6.839 | 6.132 | 0.707 | 5.958 | 0.881 |
| 23 | 5.761 | 6.102 | −0.341 | 6.057 | −0.296 |
| 24 | 5.939 | 6.008 | −0.069 | 5.888 | 0.051 |
| 25 a | 6.229 | 6.236 | −0.007 | 6.359 | −0.130 |
| 26 | 6.125 | 6.074 | 0.051 | 6.151 | −0.026 |
| 27 | 6.347 | 6.301 | 0.046 | 6.365 | −0.018 |
| 28 | 6.328 | 6.282 | 0.046 | 6.292 | 0.036 |
| 29 a | 5.138 | 5.202 | −0.064 | 5.084 | 0.054 |
| 30 | 5.147 | 5.186 | −0.039 | 5.122 | 0.025 |
| 31 | 5.148 | 5.165 | −0.017 | 5.111 | 0.037 |
| 32 | 5.155 | 5.154 | 0.001 | 5.148 | 0.007 |
| 33 | 5.182 | 5.169 | 0.013 | 5.184 | −0.002 |
| 34 a | 5.190 | 5.185 | 0.005 | 5.157 | 0.033 |
| 35 | 5.173 | 5.184 | −0.011 | 5.186 | −0.013 |
| 36 | 5.180 | 5.187 | −0.007 | 5.160 | 0.020 |
| 37 | 5.161 | 5.127 | 0.034 | 5.146 | 0.015 |
| 38 | 5.166 | 5.194 | −0.028 | 5.153 | 0.013 |
| 39 a | 5.236 | 5.222 | 0.014 | 5.233 | 0.003 |
| 40 | 5.238 | 5.284 | −0.046 | 5.234 | 0.004 |
| 41 a | 5.246 | 5.237 | 0.009 | 5.273 | −0.027 |
| 42 | 5.264 | 5.291 | −0.027 | 5.282 | −0.018 |
| 43 | 5.198 | 5.156 | 0.042 | 5.200 | −0.002 |
| 44 | 5.213 | 5.222 | −0.009 | 5.209 | 0.004 |
| 45 | 5.221 | 5.177 | 0.044 | 5.236 | −0.015 |
| 46 a | 5.228 | 5.230 | −0.002 | 5.247 | −0.019 |
| 47 | 5.281 | 5.243 | 0.038 | 5.270 | 0.011 |
| 48 | 5.312 | 5.310 | 0.002 | 5.277 | 0.035 |
| 49 | 5.309 | 5.265 | 0.044 | 5.294 | 0.015 |
| 50 | 5.311 | 5.285 | 0.026 | 5.271 | 0.040 |
| 51 a | 5.321 | 5.279 | 0.042 | 5.337 | −0.016 |
| 52 | 5.341 | 5.341 | 0.000 | 5.339 | 0.002 |
| 53 | 5.370 | 5.303 | 0.067 | 5.377 | −0.007 |
| 54 | 5.381 | 5.356 | 0.025 | 5.388 | −0.007 |
| 55 a | 5.541 | 5.626 | −0.085 | 5.603 | −0.062 |
| 56 | 5.577 | 5.640 | −0.063 | 5.574 | 0.003 |
| 57 | 5.521 | 5.594 | −0.073 | 5.565 | −0.044 |
| 58 | 5.536 | 5.656 | −0.120 | 5.569 | −0.033 |
| 59 | 5.611 | 5.633 | −0.022 | 5.638 | −0.027 |
| 60 a | 5.676 | 5.698 | −0.022 | 5.641 | 0.035 |
| 61 | 5.703 | 5.659 | 0.044 | 5.682 | 0.021 |
| 62 | 5.740 | 5.714 | 0.026 | 5.690 | 0.050 |
| 63 | 5.455 | 5.408 | 0.047 | 5.475 | −0.020 |
| 64 | 5.469 | 5.469 | 0.000 | 5.479 | −0.010 |
| 65 a | 5.476 | 5.442 | 0.034 | 5.522 | −0.046 |
| 66 | 5.491 | 5.491 | 0.000 | 5.526 | −0.035 |




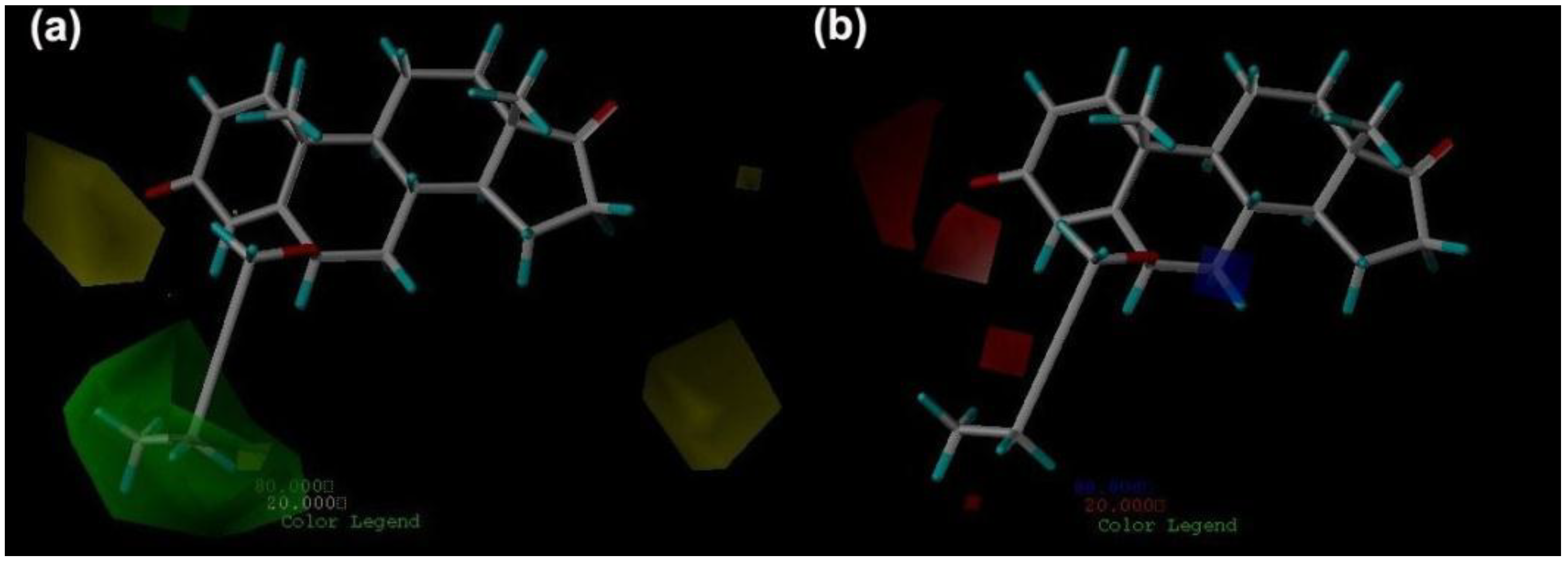
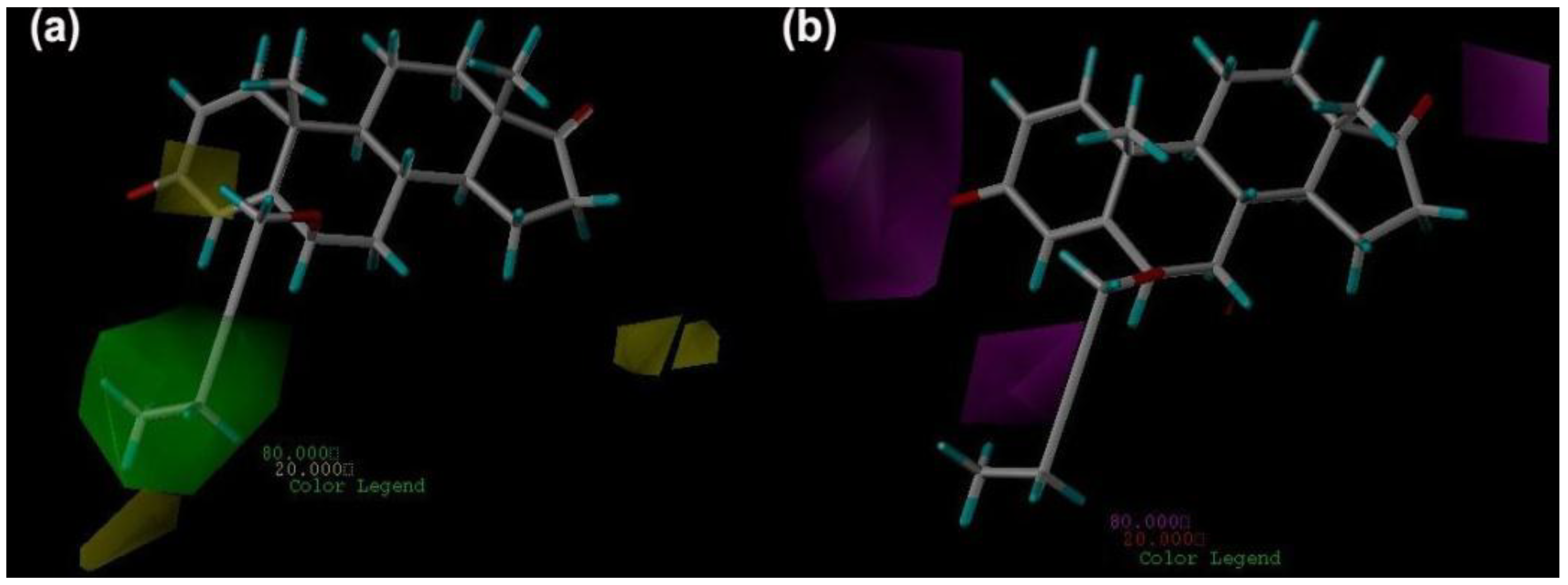
2.3. CoMFA Contour Maps

2.4. CoMSIA Contour Maps

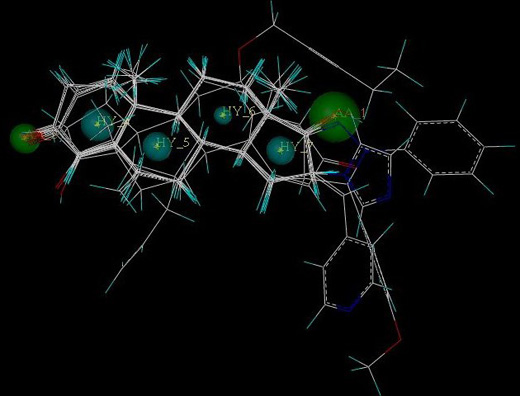
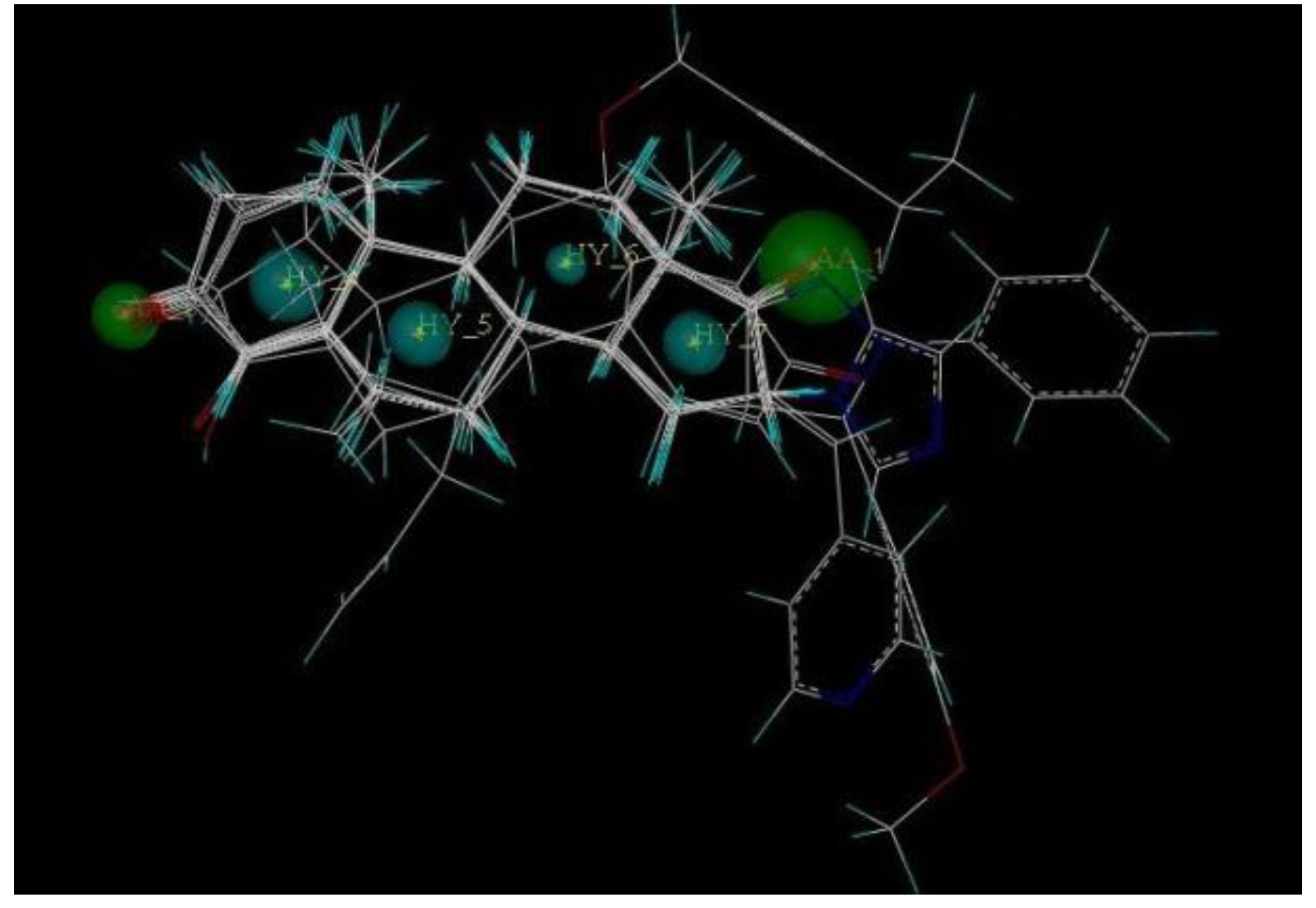
2.5. Pharmacophore Generation
| No. | Specificity | N_hits | Features | Pareto rank | Energy | Sterics | H-bond | Mol_Qry |
|---|---|---|---|---|---|---|---|---|
| Model_01 | 5.09 | 8 | 6 | 0 | 54.31 | 1158.80 | 56.90 | 17.53 |
| Model_02 | 3.67 | 8 | 6 | 0 | 20.85 | 1146.20 | 57.00 | 13.72 |
| Model_03 | 5.09 | 8 | 6 | 0 | 12657.71 | 1151.00 | 56.90 | 28.55 |
| Model_04 a | 5.15 | 8 | 6 | 0 | 8.76 | 1142.60 | 56.00 | 41.94 |
| Model_05 | 4.04 | 8 | 6 | 0 | 7.43 | 1142.30 | 55.50 | 41.25 |
| Model_06 | 4.04 | 8 | 6 | 0 | 5.39 | 1147.30 | 54.10 | 10.74 |
| Model_07 | 4.04 | 8 | 6 | 0 | 7.83 | 1135.50 | 56.60 | 35.13 |
| Model_08 | 4.04 | 8 | 6 | 0 | 8.63 | 1140.00 | 56.60 | 13.95 |
| Model_09 | 4.04 | 8 | 6 | 0 | 11.75 | 1140.40 | 56.60 | 17.87 |
| Model_10 | 4.04 | 8 | 6 | 0 | 7.23 | 1138.90 | 56.10 | 4.67 |
| Model_11 | 4.04 | 8 | 6 | 0 | 16.69 | 1146.70 | 55.50 | 41.25 |
| Model_12 | 5.09 | 8 | 6 | 0 | 24.98 | 1149.10 | 57.00 | 1.67 |
| Model_13 | 4.04 | 8 | 6 | 0 | 12.79 | 1152.80 | 56.60 | 0.00 |
| Model_14 | 4.04 | 8 | 6 | 0 | 29.82 | 1148.80 | 55.50 | 35.13 |
| Model_15 | 4.04 | 8 | 6 | 0 | 38.52 | 1158.60 | 54.50 | 11.47 |
| Model_16 | 5.15 | 8 | 6 | 0 | 7.25 | 1137.40 | 54.40 | 41.94 |
| Model_17 | 5.09 | 8 | 6 | 0 | 49.49 | 1151.90 | 56.90 | 2.81 |
| Model_18 | 3.69 | 8 | 6 | 0 | 7.07 | 1145.50 | 53.00 | 35.13 |
| Model_19 | 3.90 | 8 | 5 | 0 | 28.40 | 1150.00 | 55.90 | 10.03 |
| Model_20 | 4.04 | 8 | 6 | 0 | 9.09 | 1147.40 | 56.60 | 0.00 |

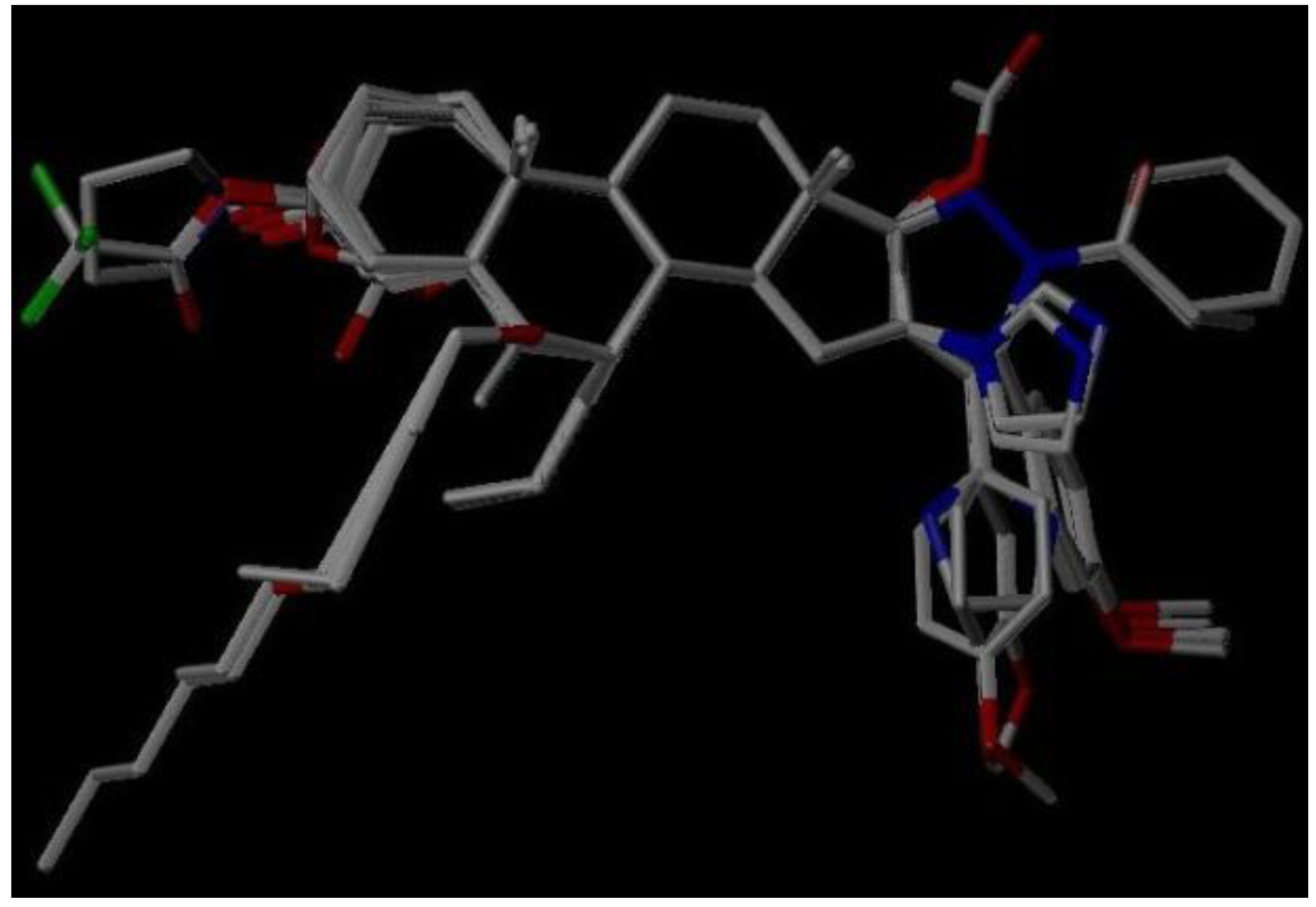
2.6. Virtual Screening
| Hit Compound | Structure | pIC50 (Predicted by CoMFA) | pIC50 (Predicted by CoMSIA) |
|---|---|---|---|
| NCI 51178 |  | 6.620 | 5.502 |
| NCI 51181 |  | 6.438 | 5.443 |
| NCI 51183 |  | 6.254 | 5.330 |
| NCI 51184 |  | 6.180 | 5.429 |
| NCI 77798 |  | 6.728 | 6.946 |
| NCI 79104 |  | 6.571 | 7.032 |
3. Experimental Section
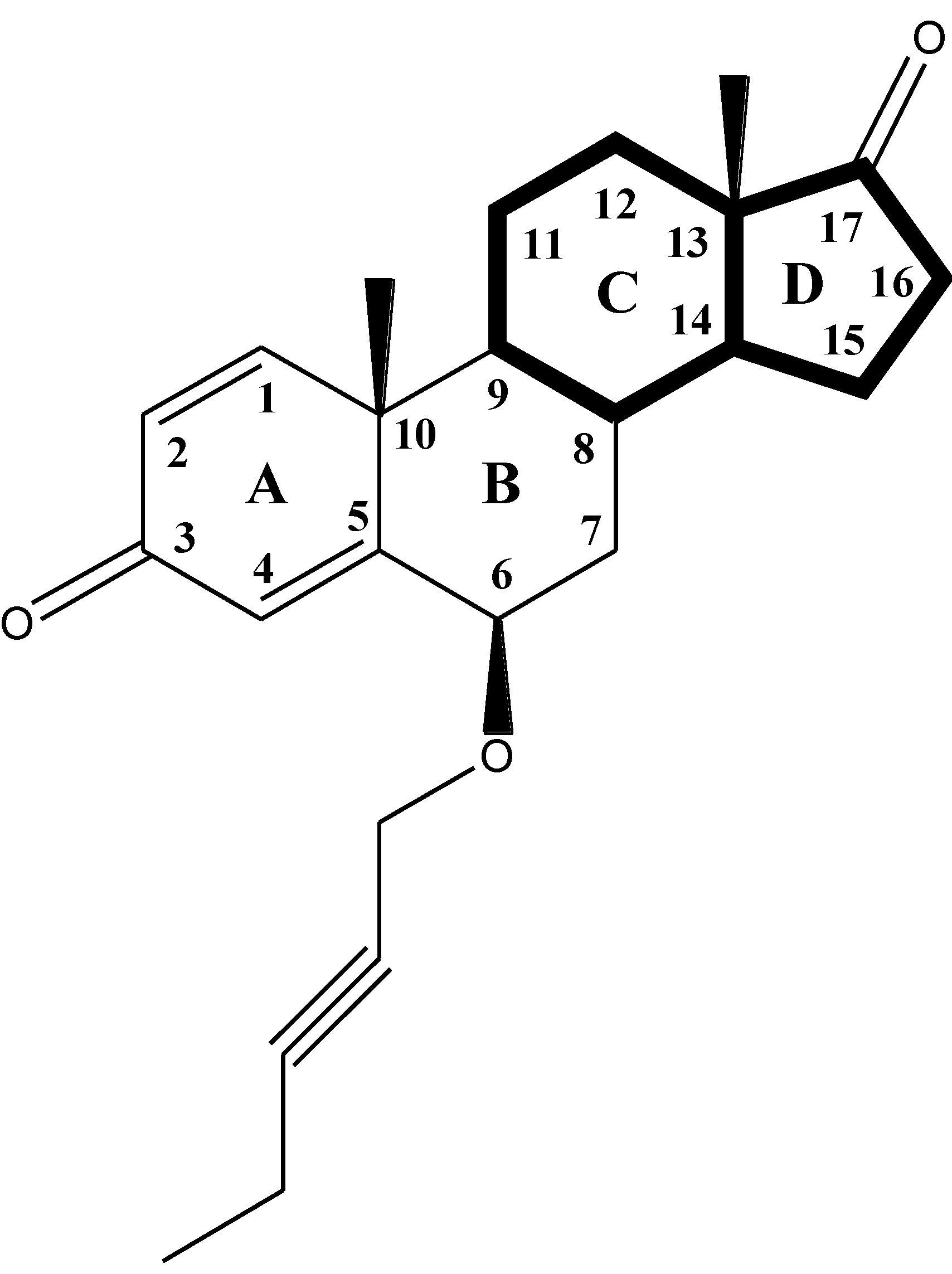
3.1. Compounds and Biological Data
3.2. Molecular Modeling and Alignment

3.3. CoMFA and CoMSIA Models
3.4. Statistical Analysis
3.5. Pharmacophore Hypothesis
4. Conclusions
Statements
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Winer, E.P.; Hudis, C.; Burstein, H.J.; Wolff, A.C.; Pritchard, K.I.; Ingle, J.N.; Chlebowski, R.T.; Gelber, R.; Edge, S.B.; Gralow, J.; et al. American society of clinical oncology technology assessment on the use of aromatase inhibitors as adjuvant therapy for postmenopausal women with hormone receptor-positive breast cancer: Status report 2004. J. Clin. Oncol. 2005, 23, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Appraising adjuvant aromatase inhibitor therapy. Oncologist 2006, 11, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; Brodie, A.M.H. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids 2007, 72, 7–25. [Google Scholar] [CrossRef] [PubMed]
- Brueggemeier, R.W.; Hackett, J.C.; Diaz-Cruz, E.S. Aromatase inhibitors in the treatment of breast cancer. Endocr. Rev. 2005, 26, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Recanatini, M.; Cavalli, A.; Valenti, P. Nonsteroidal aromatase inhibitors: Recent advances. Med. Res. Rev. 2002, 22, 282–304. [Google Scholar] [CrossRef] [PubMed]
- Seralini, G.E.; Moslemi, S. Aromatase inhibitors: Past, present and future. Mol. Cell Endocrinol. 2001, 178, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Rashid, R.; Chen, S. Binding features of steroidal and nonsteroidal inhibitors. Steroids 2011, 76, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Dutta, U.; Pant, K. Aromatase inhibitors: Past, present and future in breast cancer therapy. Med. Oncol. 2008, 25, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.S.; Mezey, P.G.; Chou, K.C. Heuristic molecular lipophilicity potential (HMLP): A 2D QSAR study to LADH of molecular family pyrazole and derivatives. J. Comput. Chem. 2005, 26, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.S.; Huang, R.B.; Wei, Y.T. Multiple field three dimensional quantitative structure-activity relationship (MF-3D QSAR). J. Comput. Chem. 2008, 29, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.S.; Huang, R.B. Review: Recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr. Protein Pept. Sci. 2008, 9, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.S.; Huang, R.B.; Wei, Y.T. Fragment-based quantitative structure-activity relationship (FB QSAR) for fragment-based drug design. J. Comput. Chem. 2009, 30, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Prado-Prado, F.J.; Gonzalez-Diaz, H.; de la Vega, O.M.; Ubeira, F.M.; Chou, K.C. Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for Input-Coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg. Med. Chem. 2008, 16, 5871–5880. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Fossa, P. Docking-based 3D QSAR analyses of pyrazole derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Mol. Model. 2012, 18, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Z.; Liu, Y.J.; Hu, S.Y.; Zhang, H.B. 3D QSAR study of Chk1 kinase inhibitors based on docking. J. Mol. Model. 2012, 18, 3669–3694. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, R.; Shen, B.Z.; Xu, Y.; Liu, Y.F.; Zheng, H.; Yao, W.B. Docking-based 3D QSAR modeling of the inhibitors of IMP metallo-β-lactamase. Med. Chem. Res. 2013, 22, 1730–1739. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Sirois, S.; Wei, D.Q.; Du, Q.S.; Chou, K.C. Virtual screening for SARS-CoV protease based on KZ7088 pharmacophore points. J. Chem. Inf. Comput. Sci. 2004, 44, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ma, Y.; Wang, R.L.; Xu, W.R.; Wang, S.Q.; Chou, K.C. Find novel dual-agonist drugs for treating type 2 diabetes by means of cheminformatics. Drug Desi. Dev. Ther. 2013, 7, 279–287. [Google Scholar]
- Bhatt, H.G.; Patel, P.K. Pharmacophore modeling, virtual screening and 3D QSAR studies of 5-tetrahydroquinolinylidine aminoguanidine derivatives as sodium hydrogen exchanger inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Thota, S.; Karkra, N.; Minu, M.; Zimmer, C.; Hartmann, R.W. Synthesis and aromatase inhibitory activity of some new 16E-arylidenosteroids. Bioorg. Chem. 2012, 45, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; Davies, H.M. Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Guleria, S.; Thota, S.; Bodhankar, S.L.; Patwardhan, M.R.; Zimmer, C.; Hartmann, R.W.; Harvey, A.L. Design, synthesis and evaluation of novel 16-imidazolyl substituted steroidal derivatives possessing potent diversified pharmacological properties. Steroids 2012, 77, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Valera, C.; Tavares da Silva, E.J.; Amaral, C.; Correia da Silva, G.; Baptista, T.; Alcaro, S.; Costa, G.; Carvalho, R.A.; Teixeira, N.A.A.; Roleira, F.M.F. New structure-activity relationships of A- and D-Ring modified steroidal aromatase inhibitors: Design, synthesis, and biochemical evaluation. J. Med. Chem. 2012, 55, 3992–4002. [Google Scholar] [CrossRef] [PubMed]
- Valera, C.L.; Amaral, C.; Correia-da-Silva, G.; Carvalho, R.A.; Teixeira, N.A.; Costa, S.C.; Roleira, F.M.F.; Tavares-da-Silva, E.J. Design, synthesis and biochemical studies of new 7α-allylandrostanes as aromatase inhibitors. Steroids 2013, 78, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.M.; Al-Omar, M.A.; Bhat, M.A.; Amr, A.E.; Al-Mohizea, A.M. Steroidal pyrazolines evaluated as aromatase and quinone reductase-2 inhibitors for chemoprevention of cancer. Int. J. Biol. Macromol. 2012, 50, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Böhm, M.; Stürzebecher, J.; Klebe, G. Three-dimensional quantitative structure-activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J. Med. Chem. 1999, 42, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U. Recent progress in CoMFA methodology and related techniques. Perspect. Drug Discov. 1998, 3, 25–39. [Google Scholar] [CrossRef]
- Dorfman, R.J.; Smith, K.M.; Masek, B.B.; Clark, R.D. A knowledge-based approach to generating diverse but energetically representative ensembles of ligand conformers. J. Comput. Aid. Mol. Des. 2008, 22, 681–691. [Google Scholar] [CrossRef]
- Caballero, J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J. Mol. Graph. Model. 2010, 29, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kothandan, G.; Madhavan, T.; Gadhe, C.G.; Cho, S.J. A combined 3D QSAR and pharmacophore-based virtual screening for the identification of potent p38 MAP kinase inhibitors: An in silico approach. Med. Chem. Res. 2013, 22, 1773–1787. [Google Scholar] [CrossRef]
- Thaimattam, R.; Daga, P.R.; Banerjee, R.; Iqbal, J. 3D QSAR studies on c-Src kinase inhibitors and docking analyses of a potent dual kinase inhibitor of c-Src and c-Abl kinases. Bioorg. Med. Chem. 2005, 13, 4704–4712. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.J.; Barbault, F.; Delamar, M.; Zhang, R.S. Receptor- and ligand-based 3D QSAR study for a series of non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009, 17, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Richmond, N.J.; Abrams, C.A.; Wolohan, P.R.N.; Abrahamian, E.; Willett, P.; Clark, R.D. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J. Comput. Aid. Mol. Des. 2006, 20, 567–587. [Google Scholar] [CrossRef]
- Shepphird, J.K.; Clark, R.D. A marriage made in torsional space: Using GALAHAD models to drive pharmacophore multiplet searches. J. Comput. Aid. Mol. Des. 2006, 20, 763–771. [Google Scholar] [CrossRef]
- Andrade, C.H.; Salum, L.B.; Pasqualoto, K.F.M.; Ferreira, E.I.; Andricopulo, A.D. Three-dimensional quantitative structure-activity relationships for a large series of potent antitubercular agents. Lett. Drug Des. Discov. 2008, 5, 377–387. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.M.; Deng, E.Z.; Lin, H.; Chou, K.C. iTIS-PseTNC: A sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal. Biochem. 2014, 462, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Deng, E.Z.; Yuan, L.F.; Liu, L.; Lin, H.; Chen, W.; Chou, K.C. iCTX-Type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. Biomed. Res. Int. 2014, 2014, 286419. [Google Scholar] [PubMed]
- Fan, Y.N.; Xiao, X.; Min, J.L.; Chou, K.C. iNR-Drug: Predicting the interaction of drugs with nuclear receptors in cellular networking. Int. J. Mol. Sci. 2014, 15, 4915–4937. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.H.; Deng, E.Z.; Xu, L.Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014, 30, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Lan, X.; Xu, R.; Zhou, J.; Wang, X.; Chou, K.C. iDNA-Prot|dis: Identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS One 2014, 9, e106691. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Xiao, X.; Lin, W.Z.; Chou, K.C. iMethyl-PseAAC: Identification of protein methylation sites via a pseudo amino acid composition approach. Biomed. Res. Int. 2014, 2014, 947416. [Google Scholar] [PubMed]
- Xu, Y.; Wen, X.; Shao, X.J.; Deng, N.Y.; Chou, K.C. iHyd-PseAAC: Predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int. J. Mol. Sci. 2014, 15, 7594–7610. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wen, X.; Wen, L.S.; Wu, L.Y.; Deng, N.Y.; Chou, K.C. iNitro-Tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS One 2014, 9, e105018. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Qiu, K.; Xie, X. 3D QSAR Studies, Pharmacophore Modeling and Virtual Screening on a Series of Steroidal Aromatase Inhibitors. Int. J. Mol. Sci. 2014, 15, 20927-20947. https://doi.org/10.3390/ijms151120927
Xie H, Qiu K, Xie X. 3D QSAR Studies, Pharmacophore Modeling and Virtual Screening on a Series of Steroidal Aromatase Inhibitors. International Journal of Molecular Sciences. 2014; 15(11):20927-20947. https://doi.org/10.3390/ijms151120927
Chicago/Turabian StyleXie, Huiding, Kaixiong Qiu, and Xiaoguang Xie. 2014. "3D QSAR Studies, Pharmacophore Modeling and Virtual Screening on a Series of Steroidal Aromatase Inhibitors" International Journal of Molecular Sciences 15, no. 11: 20927-20947. https://doi.org/10.3390/ijms151120927
APA StyleXie, H., Qiu, K., & Xie, X. (2014). 3D QSAR Studies, Pharmacophore Modeling and Virtual Screening on a Series of Steroidal Aromatase Inhibitors. International Journal of Molecular Sciences, 15(11), 20927-20947. https://doi.org/10.3390/ijms151120927