Differential Transcriptome Analysis between Paulownia fortunei and Its Synthesized Autopolyploid

Abstract
:1. Introduction
2. Results
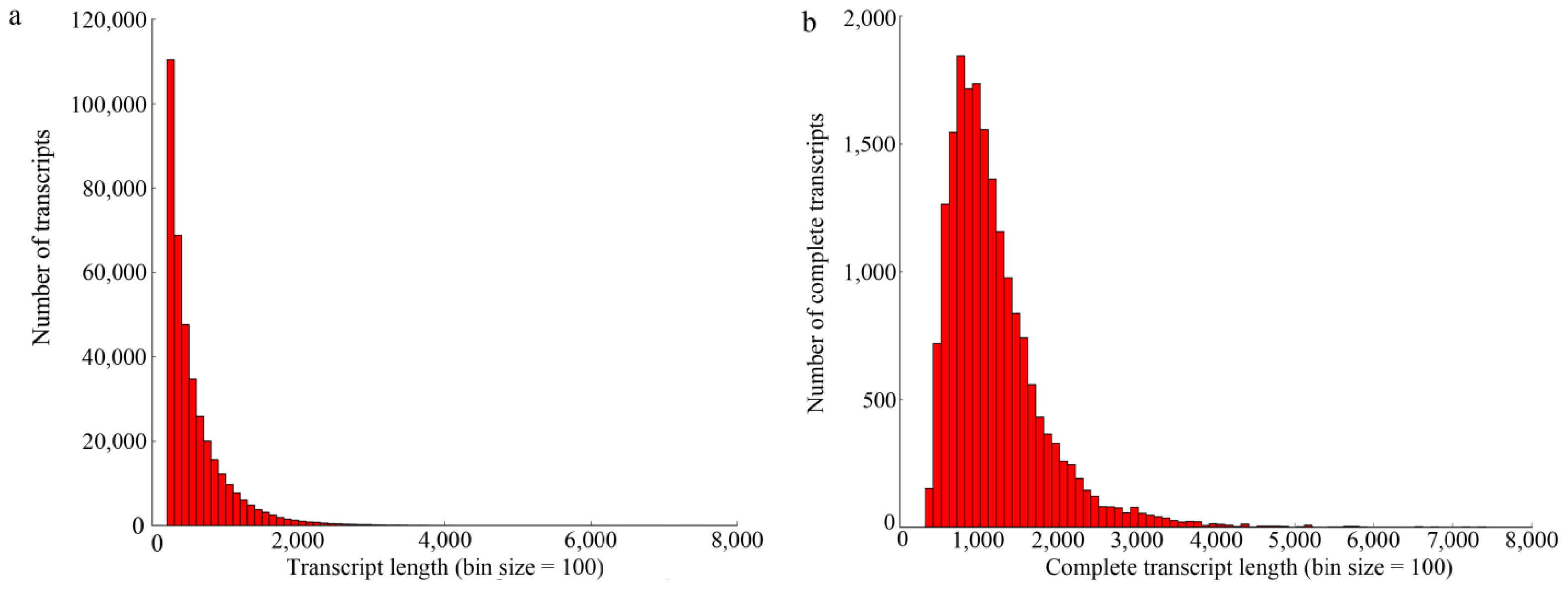
2.1. Illumina Paired-End Sequencing and De Novo Assembly
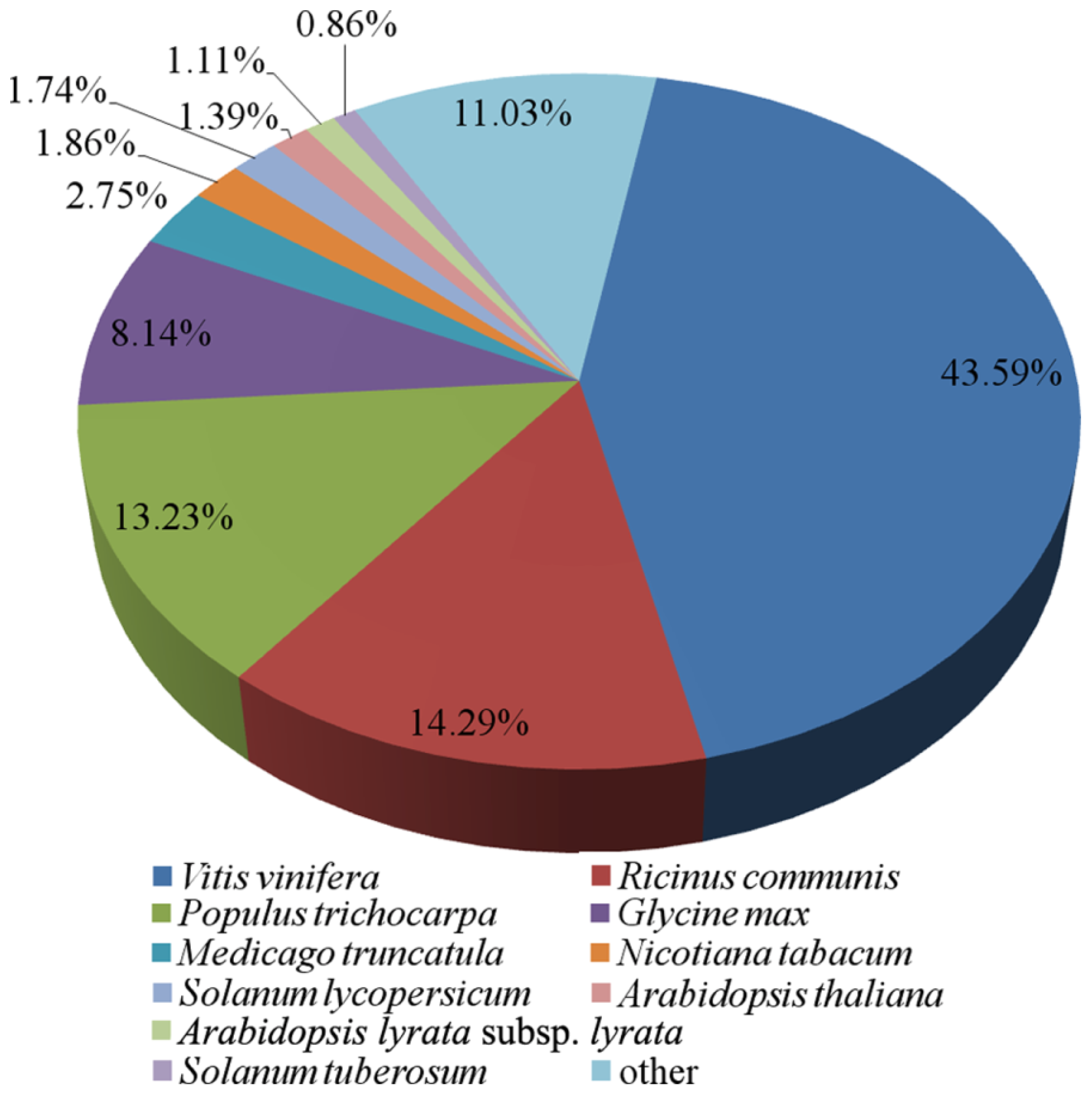
2.2. Annotation of the Predicted Complete Transcripts
2.3. Functional Classification Using GO, KOG and KEGG
2.4. Analysis of Differentially Expressed Transcripts between Diploid and Autotetraploid P. fortunei
2.5. Differentially Expressed Transcripts Related to Energy Metabolism
2.6. Transcriptomic Changes Related to Genetic Information Storage and Processing
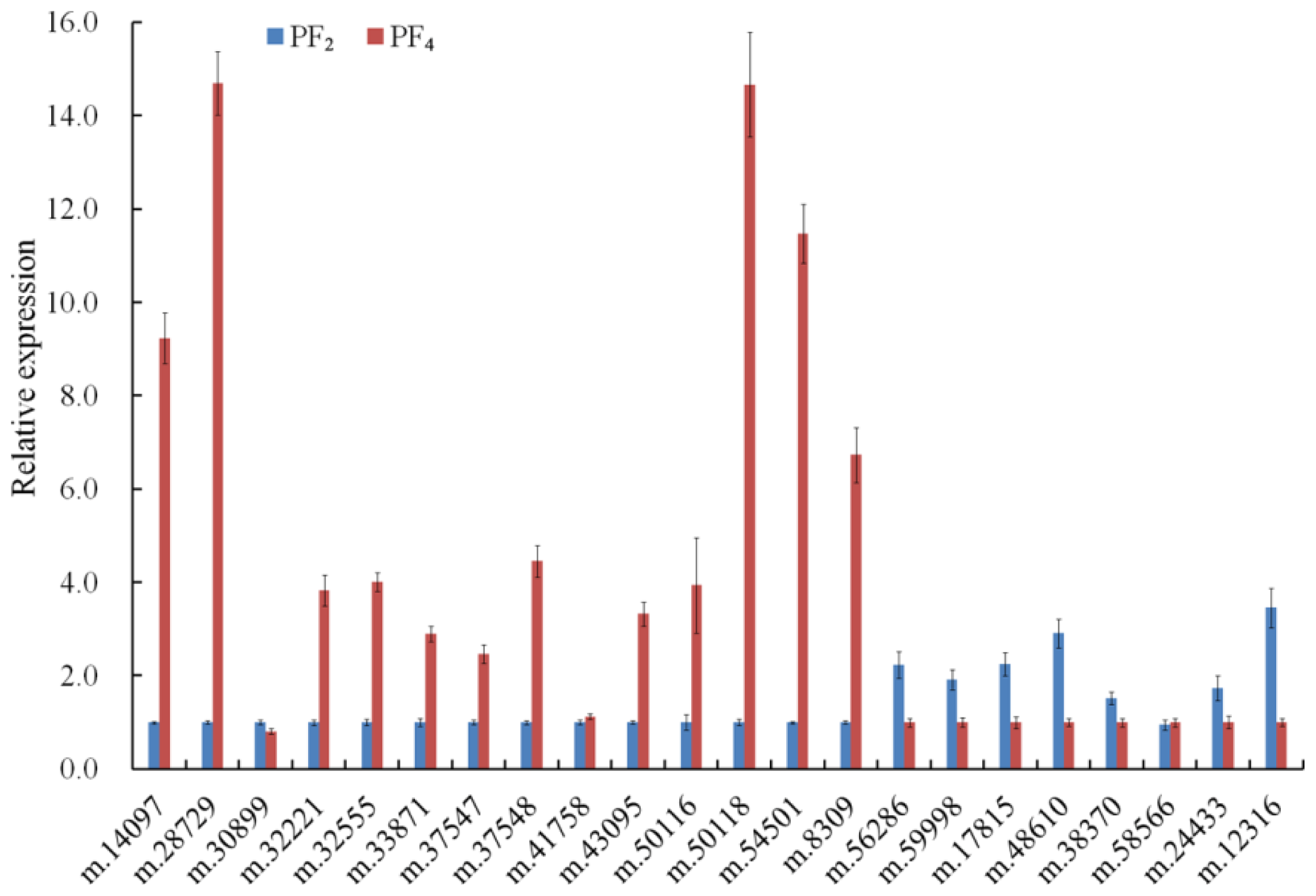
2.7. Verification of DETs by Quantitative Real-Time PCR
3. Discussion
4. Experimental Section
4.1. Tissue Collection and RNA Isolation
4.2. cDNA Library Preparation, Sequencing and De Novo Assembly
4.3. Functional Annotation and Categorization of the Transcripts
4.4. Expression Abundance Analysis
4.5. Functional Analysis of DETs
4.6. Quantitative Real-Time PCR Analysis
5. Conclusions
Supplementary Information
ijms-15-05079-s001.pdfAcknowledgments
Conflicts of Interest
References
- Caparrósa, S.; Díaza, M.J.; Arizaa, J.; Lópeza, F.; Jiménezb, L. New perspectives for Paulownia fortunei L valorisation of the autohydrolysis and pulping processes. Bioresour. Technol. 2008, 99, 741–749. [Google Scholar]
- Rai, A.K.; Singh, S.P.; Luxmi, C.; Savita, G. Paulownia fortunei—A new fiber source for pulp and paper. Indian Pulp Pap. Tech. Assoc. 2000, 12, 51–56. [Google Scholar]
- Ipekci, Z.; Gozukirmizi, N. Direct somatic embryogenesis and synthetic seed production from Paulownia elongata. Plant Cell Rep. 2003, 22, 16–24. [Google Scholar]
- Tang, R.C.; Carpenter, S.B.; Wittwer, R.F.; Graves, D.H. Paulownia—A crop tree for wood products and reclamation of surface mined land. South J. Appl. For. 1980, 4, 19–24. [Google Scholar]
- Melhuish, J.H.; Gentry, C.E.; Beckjord, P.R. Paulownia tomentosa seedling growth at differing levels of ph nitrogen and phosphorus. J. Environ. Hort. 1990, 8, 205–207. [Google Scholar]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar]
- Cifuentes, M.; Grandont, L.; Moore, G.; Chevre, A.M.; Jenczewski, E. Genetic regulation of meiosis in polyploid species: New insights into an old question. New Phytol. 2010, 186, 29–36. [Google Scholar]
- Higgins, J.; Magusin, A.; Trick, M.; Fraser, F.; Bancroft, I. Use of mRNA-seq to discriminate contributions to the transcriptome from the constituent genomes of the polyploid crop species Brassica napus. BMC Genomics 2012, 13, 247. [Google Scholar]
- Kaensaksiri, T.; Soontornchainaksaeng, P.; Soonthornchareonnon, N.; Prathanturarug, S. In vitro induction of polyploidy in Centella asiatica (L) Urban. Plant Cell Tiss. Organ Cult. 2011, 107, 187–194. [Google Scholar]
- Yu, Z.; Haberer, G.; Matthes, M.; Rattei, T.; Mayer, K.F.; Gierl, A.; Torres-Ruiz, R.A. Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 17809–17814. [Google Scholar]
- Wang, J.; Tian, L.; Madlung, A.; Lee, H.S.; Chen, M.; Lee, J.J.; Watson, B.; Kagochi, T.; Comai, L.; Chen, Z.J. Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 2004, 167, 1961–1973. [Google Scholar]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar]
- Jiang, J.; Shao, Y.; Du, K.; Ran, L.; Fang, X.; Wang, Y. Use of digital gene expression to discriminate gene expression differences in early generations of resynthesized Brassica napus and its diploid progenitors. BMC Genomics 2013, 14, 72. [Google Scholar]
- Hovav, R.; Udall, J.A.; Chaudhary, B.; Hovav, E.; Flagel, L.; Hu, G.; Wendel, J.F. The evolution of spinnable cotton fiber entailed prolonged development and a novel metabolism. PLoS Genet. 2008, 4, e25. [Google Scholar]
- Salmon, A.; Ainouche, M.L.; Wendel, J.F. Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 2005, 14, 1163–1175. [Google Scholar]
- Buggs, R.J.; Doust, A.N.; Tate, J.A.; Koh, J.; Soltis, K.; Feltus, F.A.; Paterson, A.H.; Soltis, P.S.; Soltis, D.E. Gene loss and silencing in Tragopogon miscellus (Asteraceae): Comparison of natural and synthetic allotetraploids. Heredity (Edinb.) 2009, 103, 73–81. [Google Scholar]
- Udall, J.A.; Wendel, J.F. Polyploidy and crop improvement. Crop Sci. 2006, 46, S3–S14. [Google Scholar]
- Fan, G.Q.; Cao, Y.C.; Zhao, Z.L.; Yang, Z.Q. Induction of autotetraploid of Paulownia fortunei. Sci. Silv. Sin. 2007, 43, 31–35. [Google Scholar]
- Collins, L.J.; Biggs, P.J.; Voelckel, C.; Joly, S. An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Inform. 2008, 21, 3–14. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar]
- Zhai, X.Q.; Zhang, X.S.; Zhao, Z.L.; Deng, M.J.; Fan, G.Q. Study on wood physical properties of tetraploid Paulownia fortunei. J. Henan Agric. Univ. 2012, 46, 651–654. [Google Scholar]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar]
- Liu, R.; Dong, Y.; Fan, G.; Zhao, Z.; Deng, M.; Cao, X.; Niu, S. Discovery of genes related to witches’ broom disease in Paulownia tomentosa × Paulownia fortunei by a de novo assembled transcriptome. PLoS One 2013, 8, e80238. [Google Scholar]
- Garg, R.; Patel, R.K.; Jhanwar, S.; Priya, P.; Bhattacharjee, A.; Yadav, G.; Bhatia, S.; Chattopadhyay, D.; Tyagi, A.K.; Jain, M. Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol. 2011, 156, 1661–1678. [Google Scholar]
- Wang, Y.; Zeng, X.; Iyer, N.J.; Bryant, D.W.; Mockler, T.C.; Mahalingam, R. Exploring the switchgrass transcriptome using second-generation sequencing technology. PLoS One 2012, 7, e34225. [Google Scholar]
- Liu, S.; Chen, C.; Chen, G.; Cao, B.; Chen, Q.; Lei, J. RNA-sequencing tag profiling of the placenta and pericarp of pungent pepper provides robust candidates contributing to capsaicinoid biosynthesis. Plant Cell Tissue Organ. Cult. 2012, 110, 111–121. [Google Scholar]
- Hsiao, Y.Y.; Chen, Y.W.; Huang, S.C.; Pan, Z.J.; Fu, C.H.; Chen, W.H.; Tsai, W.C.; Chen, H.H. Gene discovery using next-generation pyrosequencing to develop ESTs for Phalaenopsis orchids. BMC Genomics 2011, 12, 360. [Google Scholar]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly annotation and marker discovery. BMC Genomics 2010, 11, 180. [Google Scholar]
- Huang, L.L.; Yang, X.; Sun, P.; Tong, W.; Hu, S.Q. The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PLoS One 2012, 7, e38653. [Google Scholar]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar]
- Barrero, R.A.; Chapman, B.; Yang, Y.; Moolhuijzen, P.; Keeble-Gagnere, G.; Zhang, N.; Tang, Q.; Bellgard, M.I.; Qiu, D. De novo assembly of Euphorbia fischeriana root transcriptome identifies prostratin pathway related genes. BMC Genomics 2011, 12, 600. [Google Scholar]
- Zhou, Y.; Gao, F.; Liu, R.; Feng, J.; Li, H. De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus. BMC Genomics 2012, 13, 266. [Google Scholar]
- Harhay, G.P.; Sonstegard, T.S.; Keele, J.W.; Heaton, M.P.; Clawson, M.L.; Snelling, W.M.; Wiedmann, R.T.; van Tassell, C.P.; Smith, T.P. Characterization of 954 bovine full-CDS cDNA sequences. BMC Genomics 2005, 6, 166. [Google Scholar]
- Chen, F.; Lee, Y.; Jiang, Y.; Wang, S.; Peatman, E.; Abernathy, J.; Liu, H.; Liu, S.; Kucuktas, H.; Ke, C.; et al. Identification and characterization of full-length cDNAs in channel catfish (Ictalurus punctatus) and blue catfish (Ictalurus furcatus). PLoS One 2010, 5, e11546. [Google Scholar]
- Andreassen, R.; Lunner, S.; Hoyheim, B. Characterization of full-length sequenced cDNA inserts (FLIcs) from Atlantic salmon (Salmo salar). BMC Genomics 2009, 10, 502. [Google Scholar]
- Jackson, S.; Chen, Z.J. Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 2010, 13, 153–159. [Google Scholar]
- Anssour, S.; Krugel, T.; Sharbel, T.F.; Saluz, H.P.; Bonaventure, G.; Baldwin, I.T. Phenotypic genetic and genomic consequences of natural and synthetic polyploidization of Nicotiana attenuata and Nicotiana obtusifolia. Ann. Bot. 2009, 103, 1207–1217. [Google Scholar]
- Stupar, R.M.; Bhaskar, P.B.; Yandell, B.S.; Rensink, W.A.; Hart, A.L.; Ouyang, S.; Veilleux, R.E.; Busse, J.S.; Erhardt, R.J.; Buell, C.R.; et al. Phenotypic and transcriptomic changes associated with potato autopolyploidization. Genetics 2007, 176, 2055–2067. [Google Scholar]
- Albertin, W.; Brabant, P.; Catrice, O.; Eber, F.; Jenczewski, E.; Chevre, A.M.; Thiellement, H. Autopolyploidy in cabbage (Brassica oleracea L) does not alter significantly the proteomes of green tissues. Proteomics 2005, 5, 2131–2139. [Google Scholar]
- Cervigni, G.D.; Paniego, N.; Pessino, S.; Selva, J.P.; Diaz, M.; Spangenberg, G.; Echenique, V. Gene expression in diplosporous and sexual Eragrostis curvula genotypes with differing ploidy levels. Plant Mol. Biol. 2008, 67, 11–23. [Google Scholar]
- Wang, J.; Tian, L.; Lee, H.S.; Wei, N.E.; Jiang, H.; Watson, B.; Madlung, A.; Osborn, T.C.; Doerge, R.W.; Comai, L.; et al. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 2006, 172, 507–517. [Google Scholar]
- Lu, B.; Pan, X.; Zhang, L.; Huang, B.; Sun, L.; Li, B.; Yi, B.; Zheng, S.; Yu, X.; Ding, R.; et al. A genome-wide comparison of genes responsive to autopolyploidy in Isatis indigotica using Arabidopsis thaliana Affymetrix genechips. Plant Mol. Biol. Rep. 2006, 24, 197–204. [Google Scholar]
- Fu, W.; Ma, X.; Tang, Q.; Mo, C. Karyotype analysis and genetic variation of a mutant in Siraitia grosvenorii. Mol. Biol. Rep. 2012, 39, 1247–1252. [Google Scholar]
- Salmon, A.; Ainouche, M.L. Polyploidy and DNA methylation: New tools available. Mol. Ecol. 2010, 19, 213–215. [Google Scholar]
- Ha, M.; Lu, J.; Tian, L.; Ramachandran, V.; Kasschau, K.D.; Chapman, E.J.; Carrington, J.C.; Chen, X.; Wang, X.J.; Chen, Z.J. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl. Acad. Sci. USA 2009, 106, 17835–17840. [Google Scholar]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011, 12, 323. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Pathway entry | Pathway name | Number of DETs a | Corrected p-value |
|---|---|---|---|
| map00620 | Pyruvate metabolism | 20 | 0.008 |
| map00710 | Carbon fixation in photosynthetic organisms | 17 | 0.011 |
| map00190 | Oxidative phosphorylation | 11 | 0.023 |
| map00720 | Carbon fixation pathways in prokaryotes | 9 | 0.009 |
| map00860 | Porphyrin and chlorophyll metabolism | 9 | 0.009 |
| map00906 | Carotenoid biosynthesis | 6 | 0.020 |
| map00592 | alpha-Linolenic acid metabolism | 5 | 0.038 |
| map00920 | Sulfur metabolism | 5 | 0.034 |
| map00591 | Linoleic acid metabolism | 5 | 0.015 |
| map00670 | One carbon pool by folate | 5 | 0.015 |
| map00061 | Fatty acid biosynthesis | 4 | 0.021 |
| map00590 | Arachidonic acid metabolism | 3 | 0.015 |
| map00902 | Monoterpenoid biosynthesis | 3 | 0.012 |
| map00196 | Photosynthesis-antenna proteins | 2 | 0.038 |
| map00785 | Lipoic acid metabolism | 2 | 0.014 |
| map00253 | Tetracycline biosynthesis | 2 | 0.016 |
| Transcript ID | KEGG orthology (KO) number | KEGG descriptions | E-value | KEGG pathway no.a |
|---|---|---|---|---|
| m.14097 | K02155 | V-type H+-transporting ATPase 16 kDa proteolipid subunit | 7.0 × 10−69 | map00190 |
| m.54501 | K02147 | V-type H+-transporting ATPase subunit B | 1.0 × 10−45 | map00190 |
| m.32555 | K02154 | V-type H+-transporting ATPase subunit I | 1.0 × 10−48 | map00190 |
| m.33871 | K02145 | V-type H+-transporting ATPase subunit A | 1.0 × 10−48 | map00190 |
| m.30899 * | K02144 | V-type H+-transporting ATPase 54 kD subunit | 7.0 × 10−48 | map00190 |
| m.8309 * | K00029 | malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) | 1.0 × 10−34 | map00620, map00710 |
| m.32221 * | K00029 | malate dehydrogenase (oxaloacetate-decarboxylating) (NADP+) | 8.0 × 10−43 | map00620, map00710 |
| m.28729 | K00025 | malate dehydrogenase | 6.0 × 10−54 | map00620, map00710 |
| m.37547 | K01006 | pyruvate, orthophosphate dikinase | 6.0 × 10−46 | map00620, map00710 |
| m.37548 | K01006 | pyruvate, orthophosphate dikinase | 1.0 × 10−54 | map00620, map00710 |
| m.41758 | K00873 | pyruvate kinase | 2.0 × 10−26 | map00620, map00710 |
| m.43095 | K00873 | pyruvate kinase | 4.0 × 10−31 | map00620, map00710 |
| m.50116 | K01595 | phosphoenolpyruvate carboxylase | 7.0 × 10−79 | map00620, map00710 |
| m.50118 | K01595 | phosphoenolpyruvate carboxylase | 9.0 × 10−40 | map00620, map00710 |
| Transcript ID | Function descriptions | E-value |
|---|---|---|
| m.56286 | 5′-3′ exonuclease HKE1/RAT1 | 9.0 × 10−11 |
| m.59998 | Chromatin remodeling complex SWI/SNF, component SWI2 and related ATPases (DNA/RNA helicase superfamily) | 4.0 × 10−27 |
| m.17815 | Chromatin remodeling protein HARP/SMARCAL1, DEAD-box superfamily | 8.0 × 10−7 |
| m.48610 | Polyadenylate-binding protein (RRM superfamily) | 7.0 × 10−6 |
| m.38370 | Translation initiation factor 3, subunit c (eIF-3c) | 3.0 × 10−34 |
| m.58566 | mRNA cleavage and polyadenylation factor II complex, BRR5 (CPSF subunit) | 1.0 × 10−110 |
| m.24433 | RNA Helicase | 9.0 × 10−6 |
| m.12316 | Transcription factor containing NAC and translation elongation factor EF-Ts, N-terminal domain (TS-N) domains | 3.0 × 10−8 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, X.; Deng, M.; Fan, G. Differential Transcriptome Analysis between Paulownia fortunei and Its Synthesized Autopolyploid. Int. J. Mol. Sci. 2014, 15, 5079-5093. https://doi.org/10.3390/ijms15035079
Zhang X, Deng M, Fan G. Differential Transcriptome Analysis between Paulownia fortunei and Its Synthesized Autopolyploid. International Journal of Molecular Sciences. 2014; 15(3):5079-5093. https://doi.org/10.3390/ijms15035079
Chicago/Turabian StyleZhang, Xiaoshen, Minjie Deng, and Guoqiang Fan. 2014. "Differential Transcriptome Analysis between Paulownia fortunei and Its Synthesized Autopolyploid" International Journal of Molecular Sciences 15, no. 3: 5079-5093. https://doi.org/10.3390/ijms15035079
APA StyleZhang, X., Deng, M., & Fan, G. (2014). Differential Transcriptome Analysis between Paulownia fortunei and Its Synthesized Autopolyploid. International Journal of Molecular Sciences, 15(3), 5079-5093. https://doi.org/10.3390/ijms15035079