2. Results and Discussion
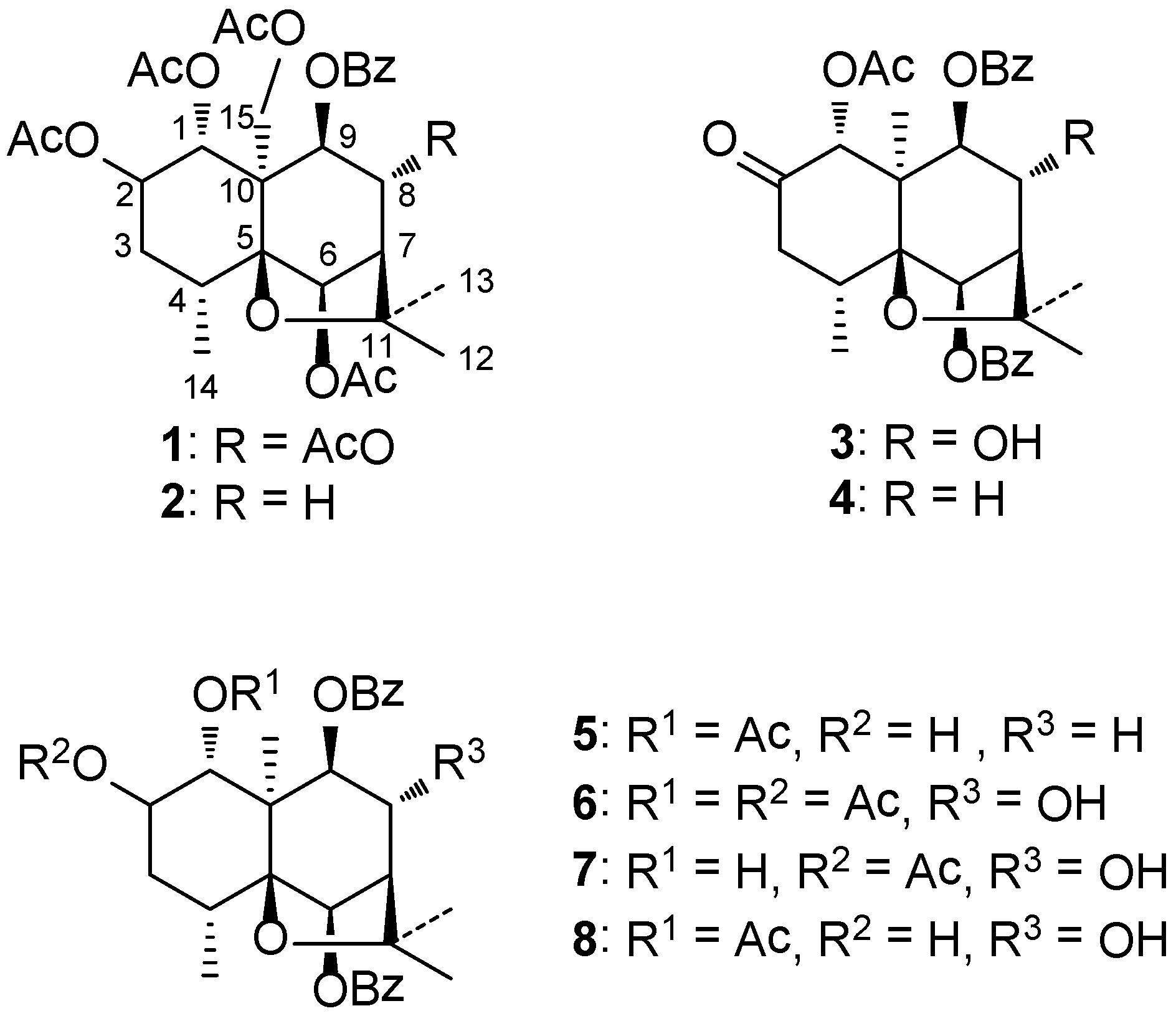
Three known compounds were identified as ejap-4 (
1) [
14], celahin B (
2) [
15,
16] and 1α-acetoxy-8α-hydroxy-6β,9β-dibenzyloxy-2-oxodihydro-β-agarofuran (
3) [
12,
13] by spectroscopic methods, but the previous reports did not confirm their absolute configurations. For this reason, a single-crystal X-ray diffraction experiment of
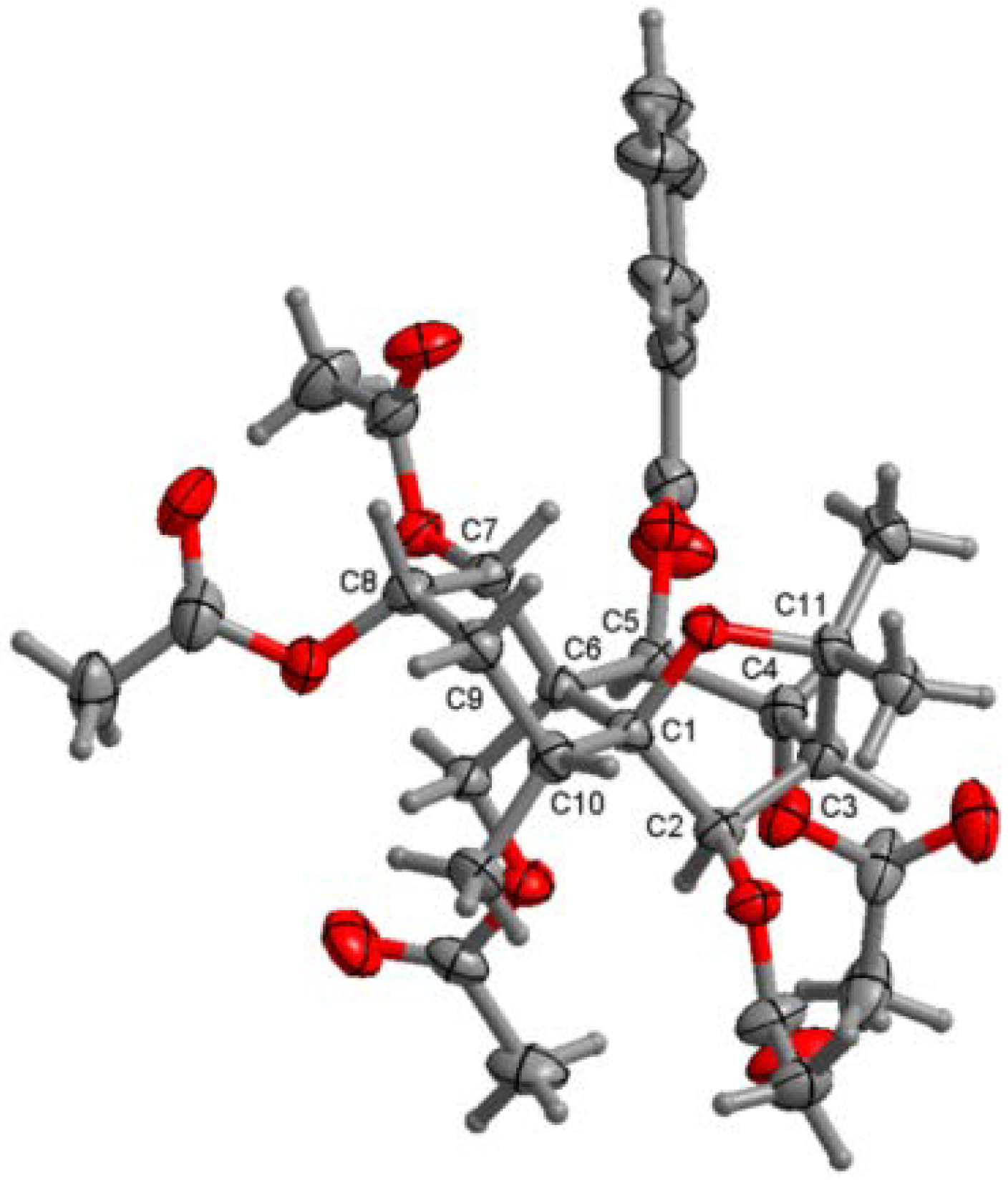
1 was performed in the present research. Its crystallographic data revealed that the torsion angle obtained corresponded to a decalin system with a
trans union between rings A and B, of which A adopted a chair conformation and B, a half-chair conformation, whereas the furan ring presented an envelope conformation (
Figure 2). Thus,
1 was elucidated as (1
R,2
S,4
R,5
S,6
R,7
R,8
R,9
R,10
S)-1,2,6,8,15-pentaacetoxy-9-benzoyloxydihydro-β-agarofuran. Taking into account biosynthetic consideration and the same NOE effects of
2 as that of
1, the absolute configuration of
2 can be proposed as 1
R,2
S,4
R,5
S,6
R,7
R,9
S,10
R because the only difference of
2 from
1 is the absence of acetoxyl group at C-8.
Figure 2.
X-ray ORTEP drawing of compound 1.
Figure 2.
X-ray ORTEP drawing of compound 1.
Compound
3 as a new compound from the same plant was reported by Wang
et al. [
12,
13], but the authors proposed an inconsistent stereochemistry for
3, due to the absence of the corresponding 2D-NMR analysis. In addition, they did not also provide the complete 1D-NMR data and other spectrometric data as well as physical properties. Therefore, the more detailed structure characterization of
3 is herein necessary.
Compound 3 was obtained as a white solid with [α]D21 +10.45° (c 0.331; CHCl3 ). The ions at m/z: 551 [M + H]+ in ESI-MS and 573.2107 [M + Na]+ in HR-ESI-MS of 3 afforded a molecular formula of C31H34O9. The IR showed the presence of hydroxyl (3493 cm−1), carbonyl (1752, 1724 cm−1) and aromatic groups (3063, 1601 cm−1).
The
13C NMR and DEPT spectra of
3 indicated 27 carbon signals comprising three ester carbonyl carbons, one ketone carbonyl carbon, one quaternary sp
3 carbon, three tertiary carbons including two ones linked to an oxygen atom, five secondary carbons including four ones linked to an oxygen atom, eight sp
2 carbons including six ones linked to a hydrogen atom, one methylene carbons, and five methyl carbons (
Table 1). Its NMR and mass spectra showed the presence of one acetate ester [δ
H 1.71 (3H, s), δ
C 20.0 (CH
3CO
2–), δ
C 169.5 (CH
3CO
2–); EI-MS
m/
z: 43 [Ac]
+] and two benzoate esters [δ
H 7.46 (2H, t,
J = 8.00 Hz), 7.49 (2H, t,
J = 8.00 Hz), 7.59 (1H, t,
J = 8.00 Hz), 7.62 (1H, t,
J = 8.00 Hz), 8.03 (2H, d,
J = 8.00 Hz), 8.05 (2H, d,
J = 8.00 Hz); δ
C 128.6 (CH), 128.7 (C), 128.8 (CH), 129.6 (CH), 129.7 (C), 129.8 (CH), 133.6 (CH), 133.8(CH)].
Except for the signals of the ester groups, 1H and 13C NMR, DEPT and HSQC spectra showed the structural characteristics of a dihydro-β-agarofuran ketone skeleton consisting of four methyl carbons [δH 1.06 (3H, d, J = 7.5 Hz)/δC 18.1, 1.46 (3H, s)/δC 20.7, 1.50 (3H, s)/δC 25.6, 1.59 (3H, s)/δC 31.2 and 1.71 (3H, s)/δC20.0], one methylene carbon [δH 2.29 (1H, d, J = 12.8 Hz) and 3.39 (1H, dd, J = 7.4, 12.8 Hz)/δC 44.0], six methine carbons with four linked to an oxygen atom [δH 2.72 (1H, s)/δC 55.7, 2.99–3.05 (m, quintet-like)/δC 38.8, δH 4.48 (1H, s)/δC 74.5, δH 4.97 (1H, s)/δC 80.5, δH 5.98 (1H, s)/δC 76.8, δH 6.27 (1H, s)/δC 76.1], three quaternary carbons with two linked to an oxygen atom [δC 54.8, 82.6, 89.8] and one ketone carbonyl carbon (δC 204.6).
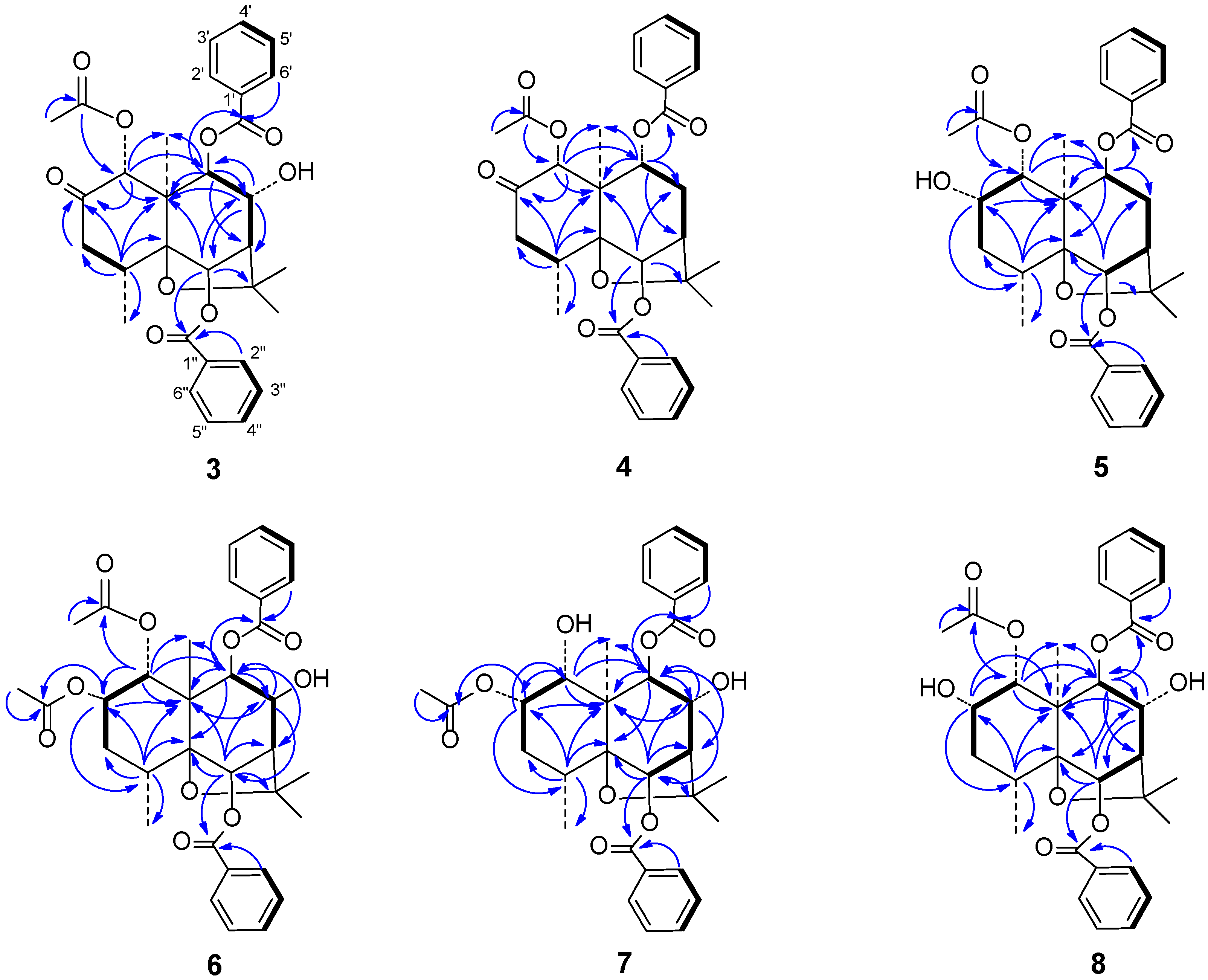
In the HMBC spectrum (
Figure 3), the correlations between δ
H 5.98 (s, H-1)/δ
C 169.5 (CH
3CO
2–), between δ
H 6.27 (s, H-6)/δ
C 165.5 (PhCO
2–), and between δ
H 4.97 (1H, s, H-9)/δ
C 165.5 (PhCO
2–) showed that the acetoxy group was at C-1 and two benzoyloxy groups were at C-6 and C-9, respectively. The correlation between δ
H 5.98 (s, H-1) and δ
C 204.6 (C=O) as well as the lowfield chemical shift and singlet of H-1 showed that the 2 position was a ketone carbonyl group. The correlations between δ
H 4.97 (s, H-9), 2.72 (s, H-7)/δ
C 74.5 (C-8) showed that the 8 site was a hydroxymethine group. The signal at δ
H 4.48 (s) was assigned to H-8 by the HSQC spectrum.
Figure 3.
Main 1H–13C long-range correlation (→) and 1H-1H correlation (–) signals in the HMBC and COSY spectra of 3–8.
Figure 3.
Main 1H–13C long-range correlation (→) and 1H-1H correlation (–) signals in the HMBC and COSY spectra of 3–8.
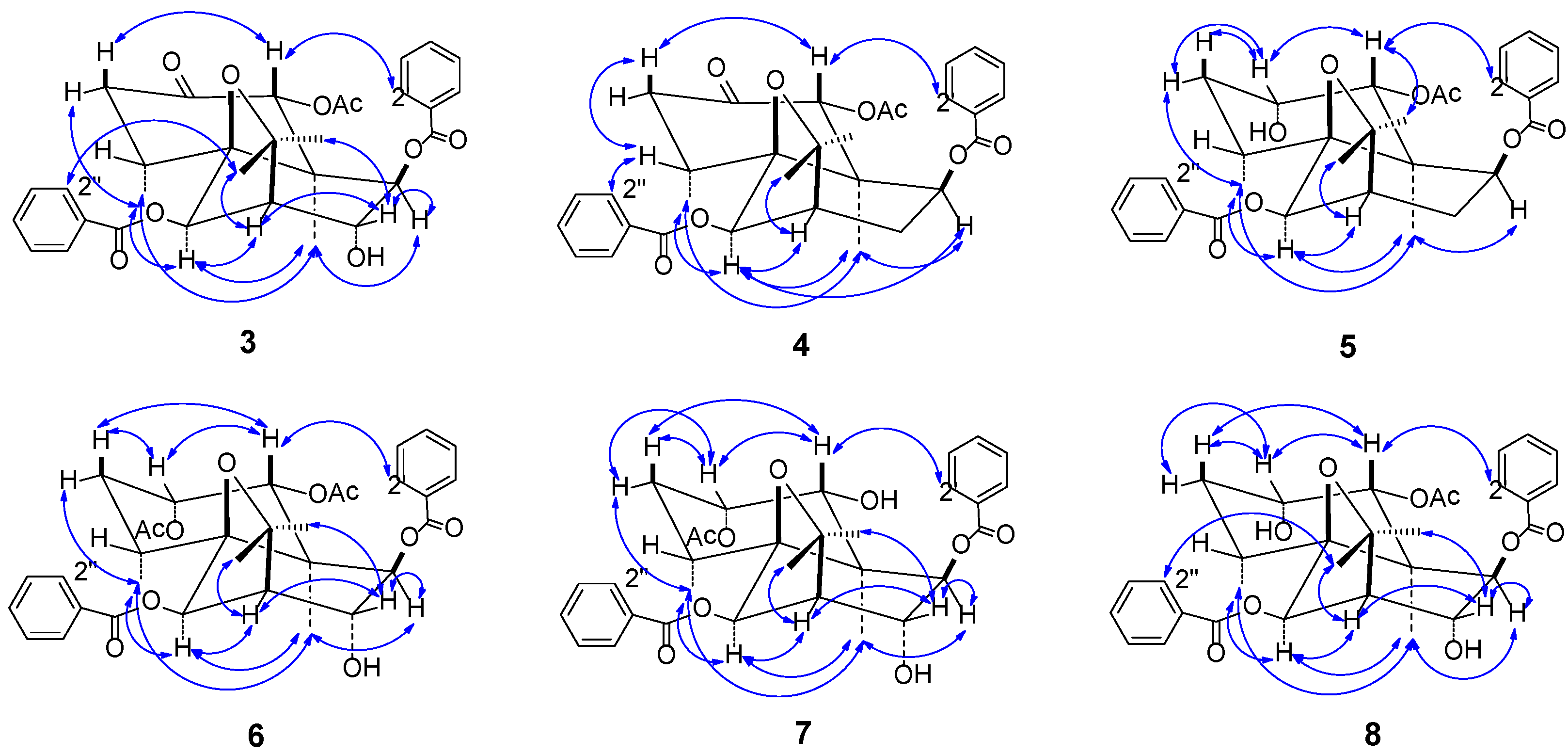
In the NOESY spectrum (
Figure 4), the correlations between δ
H 1.46 (s, H-15) and/1.06 (d,
J = 7.5 Hz, H-14), 6.27 (s, H-6), 4.97 (s, H-9) showed that 6-benzoyloxy group and 9-benzoyloxy group were equatorial and axial, respectively, and the correlations between δ
H 5.98 (s, H-1)/3.39 (dd,
J = 7.4, 12.8 Hz, H-3
ax), /8.03 (d,
J = 8.0 Hz, H-2') showed that 1-acetoxy group was equatorial. The axial assignment of 8-OH was supported by the NOE effect between δ
H 4.48 (H, s, H-8)/ 1.50 (3H, s, H-12). The above NOE cases were the same as that of
1, indicating that the space orientations of the substituents in
3 were identical to that of the substituents at the same site in
1. Thus,
3 was identified as (1
R,4
R,5
S,6
R,7
R,8
R,9
R,10
R)-1-acetoxy-6,9-dibenzoyloxy-8-hydroxydihydro-β-agarofuran-2-one.
Figure 4.
Main NOE correlation signals ( ←→ ) in the NOESY spectra of 3–8.
Figure 4.
Main NOE correlation signals ( ←→ ) in the NOESY spectra of 3–8.
Compound 4 was obtained as a white powder. Its molecular formula was established as C31H34O8 by HR-MS (m/z: 557.2144 [M + Na]+) and 13C NMR data, which had one less oxygen atom compared with that of 3. The IR spectrum showed the presence of carbonyl (1753, 1720 cm−1) and aromatic (3062, 1601 cm−1) functionalities. The UV spectrum exhibited an absorption maximum at 244, 274, 283 nm, suggesting the existence of aromatic moieties.
The
13C NMR and DEPT spectra (
Table 1 and
Table 2) indicated that
4 had one additional methylene group and one less oxygenated methine group than
3. The
1H and
13C NMR, EI-MS, IR and UV spectra displayed the presence of one acetate ester, two benzoate esters and one ketone carbonyl group. The
1H and
13C NMR spectra of
4 were similar to those of
3 except that the methylene group at C-8 of
4 replaced the oxygenated methine at C-8 of
3. Thus,
4 was identified as a 1,6,9-trisubstituted dihydroagarofuran-2-one, which was further supported by the
1H-
1H COSY and HMBC spectra. The complete assignments of the protonated carbons were made from the HSQC spectrum.
The carbonyl signals and regiosubstitution of three ester groups were assigned by HMBC (
Figure 2). The correlations between δ
C 165.6 (PhCO
2–)/δ
H 5.63 (s, H-6), between δ
C 165.2 (PhCO
2–)/δ
H 5.08 (1H, d,
J = 7.2 Hz, H-9),/δ
H 8.05 (d,
J = 7.4 Hz, benzoyl-H-2′), and between δ
C 169.5 (CH
3CO
2–)/δ
H 6.01 (s, H-1), /δ
H 1.73 (s, CH
3CO
2–) suggested that the acetyl ester and the two benzoyl esters were positioned at C-1, C-6 and C-9, respectively.
Table 1.
13C NMR and DEPT data of compounds 3–8 (100 MHz).
Table 1.
13C NMR and DEPT data of compounds 3–8 (100 MHz).
| Number | 3, δC a | 4, δC a | 5, δC | 6, δC | 7, δC | 8, δC |
|---|
| 1 | 76.8, CH | 77.5, CH | 75.7, CH | 73.0, CH | 69.2, CH | 75.7, CH |
| 2 | 204.6, C | 204.9, C | 69.1, CH | 70.9, CH | 75.1, CH | 69.0, CH |
| 3 | 44.0, CH2 | 43.9, CH2 | 34.1, CH2 | 32.1, CH2 | 32.2, CH2 | 34.2, CH2 |
| 4 | 38.8, CH | 38.9, CH | 35.9, CH | 35.8, CH | 35.5, CH | 35.8, CH |
| 5 | 89.8, C | 89.0, C | 91.9, C | 92.2, C | 92.5, C | 92.8, C |
| 6 | 76.1, CH | 80.0, CH | 81.2, CH | 77.3, CH | 77.7, CH | 77.5, CH |
| 7 | 55.7, CH | 49.0, CH | 50.4, CH | 57.6, CH | 57.7, CH | 57.7, CH |
| 8 | 74.5, CH | 32.5, CH2 | 32.5, CH2 | 75.2, C | 75.3, C | 75.3, C |
| 9 | 80.5, CH | 73.3, CH | 75.1, CH | 80.6, CH | 80.1, CH | 81.1, CH |
| 10 | 54.8, C | 55.9, C | 51.3, C | 50.5, C | 51.8, C | 50.6, C |
| 11 | 82.6, C | 83.7, C | 84.1, C | 83.4, C | 83.1, C | 83.1, C |
| 12 | 25.6, CH3 | 26.0, CH3 | 26.5, CH3 | 26.1, CH3 | 26.1, CH3 | 26.1, CH3 |
| 13 | 31.2, CH3 | 30.7, CH3 | 31.3, CH3 | 31.8, CH3 | 32.0, CH3 | 31.9, CH3 |
| 14 | 18.1, CH3 | 18.2, CH3 | 19.6, CH3 | 19.0, CH3 | 19.1, CH3 | 19.4, CH3 |
| 15 | 20.7, CH3 | 20.0, CH3 | 21.4, CH3 | 20.9, CH3 | 19.9, CH3 | 21.6, CH3 |
| AcO-1 | 20.0, CH3
169.5, C | 20.0, CH3
169.5, C | 20.9, CH3
172.2, C | 20.6, CH3
171.7, C | - | 20.9, CH3
172.2, C |
| AcO-2 | - | - | - | 21.2, CH3
172.0, C | 21.5, CH3
172.6, C | - |
| 1' | 128.7, C | 129.2, C | 130.6, CH | 131.4, C | 131.8, C | 131.5, C |
| 2'/6' | 129.8, CH | 129.8, CH | 131.1, CH | 131.2, CH | 130.8, CH | 131.1, CH |
| 3'/5' | 128.6, CH | 128.5, CH | 129.5, CH | 129.5, CH | 129.9, CH | 129.5, CH |
| 4' | 133.8, CH | 133.6, CH | 134.4, CH | 134.6, CH | 134.8, CH | 134.4, CH |
| 1'' | 129.7, C | 129.6, C | 131.3, C | 131.4, C | 131.5, C | 130.7, C |
| 2''/6'' | 129.6, CH | 129.5, CH | 131.0, C | 130.5, CH | 130.5, CH | 130.5, CH |
| 3''/5'' | 128.8, CH | 128.8, CH | 129.9, CH | 129.9, CH | 129.6, CH | 129.9, CH |
| 4'' | 133.6, CH | 133.5, CH | 134.7, CH | 134.7, CH | 134.6, CH | 134.6, CH |
| CO2-6 | 165.5, C | 165.6, C | 167.0, C | 166.8, C | 166.8, C | 166.8, C |
| CO2-9 | 165.5, C | 165.2, C | 166.8, C | 166.4, C | 166.7, C | 166.5, C |
In the NOESY spectrum (
Figure 3), the correlations between H-15 (δ
H 1.34, s)/H-9 (δ
H 5.08, d,
J = 7.2 Hz), H-6 (δ
H 5.63, s)/H-14 (δ
H 1.02, d,
J = 7.5 Hz), between H
ax-3 (δ
H 3.40, dd,
J = 7.5, 12.5 Hz)/H-1 (δ
H 6.01), and between H-6 (δ
H 5.63, s)/H-7 (δ
H 2.49, t,
J = 3.0 Hz), /H
ax-8 (δ
H 2.58, dddd,
J = 3.0, 7.2, 16.5 Hz) showed that the acetyl group and the benzoate ester at the 6 position were equatorial, while Me-14, Me-15 and the benzoate ester at the 9 position were axial. These results were the same as that of
3. Taking into account biosynthetic relationship between
4 and
3,
4 was elucidated as (1
R,4
R,5
S,6
R,7
R,9
S,10
R)-1-acetoxy-6,9-dibenzoyloxydihydro-β-agarofuran-2-one because the only difference of
4 from
3 is the absence of one hydroxyl group at the 8 position. The findings of
3 and
4 have a special significance; up to date, there have been few of examples of natural dihydro-β-agarofuran ketones.
Compound 5 was isolated as a white solid, [α]D15 +43.34° (c 2.0, CH3OH). Its molecular formula was established as C31H36O8 by HR-MS (m/z: 559.2316 [M + Na]+) and 13C NMR data, which only has two additional hydrogen atoms than that of 4. The IR spectrum showed the presence of hydroxyl (3530, 3063 cm−1), carbonyl (1718 cm−1) and aromatic (3063, 3016, 1602, 1585, 1480 cm−1) functionalities. The 1H and 13C NMR spectra of 5 were similar to those of 4. Compared with 4, 5 only had one additional oxygenated methine group and one less ketone carbonyl group.
The regiosubstitution of three ester groups were determined by HMBC experiment (
Figure 2). The correlations between δ
C 167.0 (PhCO
2–)/δ
H 5.73 (s, H-6), 8.07 (d,
J = 7.4 Hz, benzoyl-H-2''), between δ
C 166.8 (PhCO
2–)/δ
H 5.02 (1H, d,
J = 7.2 Hz, H-9), /8.03 (d,
J = 7.4 Hz, benzoyl-H-2'), and between δ
C 172.2 (MeCO
2–)/δ
H 5.44 (1H, d,
J = 3.4 Hz, H-1), /1.69 (s, CH
3CO
2–) suggested that one acetyl ester and two benzoyl esters were positioned at C-1, C-6 and C-9, respectively. In the
1H-
1H COSY spectrum, the correlation between δ
H 4.42 (1H, dd,
J = 3.0, 6.0 Hz, H-2)/δ
H 5.44 (1H, d,
J = 3.4 Hz, H-1) revealed that the only hydroxyl group was at 2 position. The results above were further supported by the upfield chemical shifts of δ
H-1, δ
H-3, δ
H-4, δ
C-1, δ
C-3 and δ
C-4 in
5 relative to those in
4. Thus,
5 was identified as the derivative of ketone carbonyl reduction of
4.
NOE correlations between H-15 (δH 1.62, s)/H-14 (δH 1.32, d, J = 7.6 Hz), /H-6 (δH 5.73, s), /H-9 (δH 5.02, d, J = 6.8 Hz), between H-1 (δH 5.44, d, J = 3.4 Hz)/H-12 (δH 1.48, s), /Hax-3 (δH 2.34, dddd, J = 3.4, 6.0, 14.4 Hz) showed that the acetyl group at 1 position and the benzoate ester at 6 position were equatorial, while Me-14, Me-15 and the benzoate ester at 9 position were axial. NOE correlations between H-2 (δH 4.42, q, J = 3.0 Hz)/H-1 (δH 5.44, d, J = 3.4 Hz), /Hax-3 (δH 2.34, dddd, J = 3.4, 6.0, 14.4 Hz) showed that 2-OH was axial. As a result, compound 5 was elucidated as (1R,2S,4R,5S,6R,7R,9S,10R)-1-acetoxy-6,9-dibenzoyloxy-2-hydroxydihydro-β-agarofuran.
Compound
6 was isolated as a white powder, [α]
D14.1 +11.63° (c 2.0, CH
3OH), was assigned the molecular formula C
33H
38O
10, as deduced from HR-MS data (
m/
z: 617.2377 [M + Na]
+) and
13C NMR data. Its IR absorptions were indicative of the presence of hydroxyl (3511 cm
−1), aromatic (3062, 3032, 1602, 1585 cm
−1) and ester (1746 and 1722 cm
−1) groups. The
1H and
13C NMR, EI-MS, IR and UV spectra displayed the presence of two acetate esters, two benzoate esters and one hydroxyl group. The
1H and
13C NMR spectra of
6 (
Table 1 and
Table 2) were similar to those of
5 except for one additional oxygenated methine group [δ
C 75.2; δ
H 4.20 (1H, d,
J = 3.2 Hz)], one additional acetate ester [δ
C 21.2, 172.0; δ
H 1.92 (s)] and one less methylene.
In the HMBC spectrum (
Figure 2), the correlations between δ
H 5.42 (1H, q,
J = 3.2 Hz, H-1), 1.49 (3H, s, AcO-1)/δ
C 171.7, between δ
H 5.49 (1H, dd,
J = 3.2, 6.4 Hz, H-2), 1.92 (3H, s, AcO-2)/δ
C 172.0, between δ
H 6.30 (s, H-6), 7.97 (d,
J = 7.4 Hz, benzoyl-H-2'')/δ
C 167.0 (PhCO
2–), between δ
H 4.93 (1H, s, H-9), 7.91 (d,
J = 7.4 Hz, benzoyl-H-2)/δ
C 166.8 (PhCO
2–) revealed that two acetyl esters were located at C-1 and C-2, respectively, while two benzoyl esters were positioned at C-6 and C-9, respectively. The correlations between δ
H 4.93 (1H, s, H-9)/δ
C 75.2 (C-8) in the HMBC and the coupling between δ
H 2.46 (1H, d,
J = 3.2 Hz, H-7) and δ
H 4.20 (1H, d,
J = 3.2 Hz, H-8) in the
1H-
1H COSY spectrum showed that the hydroxyl group was at 8 position.
Based on NOE effects (
Figure 3) and the splitting patterns and coupling constants of H-1 (δ
H 5.52, d,
J = 3.2 Hz), H-2 (δ
H 5.59, dd,
J = 3.2, 6.4 Hz), H-6 (δ
H 6.40, s) and H-9 (δ
H 5.03, s), the stereochemical assignments of C-1, C-2, C-4, C-6 and C-9 were in agreement with the stereochemistry observed at these positions in
5. NOE effect between H-8 (δ 4.30, d,
J = 3.2 Hz)/H-12 [δ
H 1.36 (3H, s)] showed that the hydroxyl group at the 8 position was axial. Thus,
6 was elucidated as (1
R,2
S,4
R,5
S,6
R,7
R,8
R,9
R,10
R)-1,2-diacetoxy-6,9-dibenzoyloxy-8-hydroxydihydro-β-agarofuran.
Compound
7 was isolated as a white powder, [α]
D15.2 +8.50° (
c 2.0, CH
3OH), possessed a molecular formula C
31H
36O
9, as deduced from HR-MS data (
m/
z: 575.2269 [M + Na]
+) and
13C NMR data. The IR spectrum showed the presence of hydroxyl (3503 cm
−1), carbonyl (1721 cm
−1) and aromatic (3016, 3010, 1602, 1584 cm
−1) functionalities. The
1H NMR,
13C NMR, EIMS, IR and UV spectra displayed the presence of one acetate ester, two benzoate esters and two hydroxyl groups. The
1H and
13C NMR spectra (
Table 1 and
Table 2) of
7, including chemical shifts, splitting patterns and coupling constants, were very similar to those of
6 except for the lack of signals of one acetyl group. In the HMBC spectrum, the correlations between δ
H 5.20 (dd,
J = 3.3, 6.0 Hz, H-2), 1.97 (s, Ac-H)/δ
C 172.6 (CH
3CO
2–), between δ
H 6.35 (s, H-6)/δ
C 166.8 (PhCO
2–), and between δ
H 4.98 (s, H-9)/δ
C 166.7 (PhCO
2–) showed that the acetyl ester and two benzoyl esters were located at C-2, C-6 and C-9, respectively. The splitting patterns and coupling constants between δ
H 4.45 (d,
J = 3.3 Hz, H-1) and 5.25 (dd,
J = 3.3, 6.0 Hz, H-2), and between δ
H 4.31 (d,
J = 3.2 Hz, H-8) and 2.50 (d,
J = 3.2 Hz, H-7) in the
1H NMR spectrum showed that two hydroxyl groups were located at C-1 and C-8, respectively. The above results were further confirmed by HMBC spectrum (
Figure 1). In the NOESY spectrum, the NOEs were observed to be exactly the same as those of
6, showing that
7 had the same stereochemistry as
6. Thus,
7 was proposed as (1
R,2
S,4
R,5
S,6
R,7
R,8
R,9
R,10
S)-2-acetoxy-6,9-dibenzoyloxy-1,8-dihydroxydihydro-β-agarofuran.
Compound
8 was isolated as a white solid [
α]
D15 +11.80° (
c 2.0, CH
3OH) and had the molecular formula C
31H
36O
9, as deduced from HR-ESI-MS data (
m/
z: 575.2271 [M + Na]
+), which was identical to that of
7. The IR spectrum showed the presence of hydroxyl (3520 cm
−1), aromatic (3063, 1602, 1585 cm
−1) and carbonyl (1746, 1722 cm
−1) groups. The
1H NMR (
Table 2),
13C NMR (
Table 1), EI-MS, IR and UV spectra displayed the presence of one acetate ester, two benzoate esters and two hydroxyl groups, in agreement with the case of
7. The
1H and
13C NMR spectra of
8 (
Table 1 and
Table 2) were nearly the same as those of
7 except for the signals of H-1 and H-2, showing that
8 and
7 had the same dihydro-β-agarofuran core, and their structural difference only lied in the substituted patterns of C-1 and C-2. Based on the splitting patterns and coupling constants in the
1H NMR of
8, the signals at δ
H 5.37 (1H, d,
J = 3.2 Hz) and 4.42 (1H, d,
J = 3.2, 6.0 Hz) were assigned as H-1 and H-2, respectively. In the HMBC spectrum, the correlations between δ
H 5.37 (H-1), 1.69 (3H, s, Ac-H)/δ
C 172.2 (CH
3CO
2–) showed that the acetoxy group was located at one site.
In the NOESY spectrum, the NOEs of
8 were observed to be the same as those of
7 (
Figure 2). As the only difference of
8 from
7 was the transfer of the acetoxy group at the 2 site in
7 to the 1 site in
8, this compound lastly was identified as having the same stereochemistry as
7. Accordingly, the structure of
8 was deduced as (1
R,2
S,4
R,5
S,6
R,7
R,8
R,9
R,10
S)-1-acetoxy-6,9-dibenzoyloxy-2,8-dihydroxydihydro-β-agarofuran.
To explore the potential bioactivities of the isolates from
P. wightiana Wall, the compounds
2–
8 were evaluated for
in vitro cytotoxic properties against HL-60, SMMC-7721, A549, MCF-7 and SW480 cell lines by MTT assays. Because of poor solubility, compound
1 was not tested for cytotoxicity. The cytotoxicity data are shown in
Table 3. The anticancer agent
cis-diaminodichloroplatinum was used as a positive control.
All the test compounds showed the definite activities in varying degrees against all tested cell lines at the concentration of 40 μM. For most of the cell lines, compounds 3, 5–8 exhibited moderate activities with IC50 values ranging from 11.8 to 30.6 μM, while 2 and 4 displayed poor activities with IC50 values >40 μM in most cases. It was worth noting that 4 showed higher activity against A-549 (IC50 = 17.4 μM). For each of the tested cell lines, 5, 6 or 7 were the most cytotoxic.
Table 2.
1H NMR data of compounds 3–8 (400 MHz; 3, 4, CD3Cl3; 5–8, CD3OD).
Table 2.
1H NMR data of compounds 3–8 (400 MHz; 3, 4, CD3Cl3; 5–8, CD3OD).
| Number | 3, δH a, J in Hz | 4, δH a, J in Hz | 5, δH, J in Hz | 6, δH, J in Hz | 7, δH, J in Hz | 8, δH, J in Hz |
|---|
| 1 | 5.98, s | 6.01, s | 5.44, d, 3.4 | 5.42, d, 3.2 | 4.45, d, 3.3 | 5.37, d, 3.2 |
| 2 | | | 4.42, dd, 3.0, 6.0 | 5.49, dd, 3.2, 6.4 | 5.20, dd, 3.3, 6.0 | 4.42, dd, 3.2, 6.0 |
| 3eq | 2.29, d, 12.8 | 2.27, d, 12.5 | 1.88, br d, 14.4 | 1.75, br d, 15.2 | 1.82, br d, 15.0 | 1.86, br d, 14.8 |
| 3ax | 3.39, dd,
7.4, 12.8 | 3.40, dd,
7.3, 12.5 | 2.34, dddd,
3.4, 6.0, 14.4 | 2.35, dddd,
3.6, 6.4, 15.1 | 2.31, dddd,
3.3, 6.0, 15.0 | 2.33, dddd,
3.2, 6.0, 14.8 |
| 4 | 2.99–3.05
quintet-like | 2.98–3.04
quintet-like | 2.48–2.55
quintet-like | 2.43–2.51
quintet-like | 2.40–2.48
quintet-like | 2.46–2.53
quintet-like |
| 6 | 6.27, s | 5.63, s | 5.73, s | 6.30, s | 6.35, s | 6.40, s |
| 7 | 2.72, s | 2.49, t, 3.0 | 2.41, t-like, 2.8 | 2.46, d, 3.2 | 2.50, d, 3.2 | 2.53, d, 3.2 |
| 8eq | 4.48, s | 2.33, dd,
3.0, 16.5 | 2.21, dd,
3.0, 16.4 | 4.20, d, 3.2 | 4.31, d, 3.2 | 4.29, d, 3.2 |
| 8ax | - | 2.58, dddd,
3.0, 7.2, 16.5 | 2.58, dddd,
3.2, 6.8, 16.4 | - | - | - |
| 9 | 4.97, s | 5.08, d, 7.2 | 5.02, d, 6.8 | 4.93, s | 4.98, s | 5.04, s |
| 12 | 1.50, s | 1.55, s | 1.45, s | 1.36, s | 1.39, s | 1.44, s |
| 13 | 1.59, s | 1.55, s | 1.48, s | 1.45, s | 1.48, s | 1.54, s |
| 14 | 1.06, d, 7.5 | 1.02, d, 7.5 | 1.32, d, 7.6 | 1.18, d, 7.6 | 1.15, d, 7.6 | 1.30, d, 7.6 |
| 15 | 1.46, s | 1.34, s | 1.62, s | 1.60, s | 1.55, s | 1.75, s |
| AcO-1 | 1.71, s | 1.73, s | 1.69, s | 1.49, s | - | 1.69, s |
| AcO-2 | - | - | - | 1.92, s | 1.97, s | - |
| 2'/6' | 8.03, d, 8.0 | 8.05, d, 7.4 | 8.03, d, 7.4 | 7.91, d, 7.4 | 8.03, d, 7.4 | 8.02, d, 7.3 |
| 3'/5' | 7.46, t, 8.0 | 7.45, t, 7.4 | 7.47, d, 7.4 | 7.37, t, 7.4 | 7.44, d, 7.4 | 7.47, d, 7.3 |
| 4' | 7.59, t, 8.0 | 7.57, t, 7.4 | 7.60, d, 7.4 | 7.51, t, 7.4 | 7.55, t, 7.4 | 7.61, d, 7.3 |
| 2''/6'' | 8.05, d, 8.0 | 8.06, d, 7.4 | 8.07, d, 7.4 | 7.97, d, 7.4 | 8.01, d, 7.4 | 8.06, d, 7.3 |
| 3''/5'' | 7.49, t, 8.0 | 7.50, t, 7.4 | 7.54, d, 7.4 | 7.44, t, 7.4 | 7.47, d, 7.4 | 7.54, d, 7.3 |
| 4'' | 7.62, t, 8.0 | 7.63, t, 7.4 | 7.66, d, 7.4 | 7.56, t, 7.4 | 7.59, t, 7.4 | 7.66, d, 7.3 |
Table 3.
Cytotoxicities of compounds 2–8 against five cancer cell lines.
Table 3.
Cytotoxicities of compounds 2–8 against five cancer cell lines.
| Compound | IC50 (μM) |
|---|
| HL-60 | SMMC-7721 | A-549 | MCF-7 | SW480 |
|---|
| 2 | >40 | >40 | >40 | >40 | >40 |
| 3 | 17.6 | 20.8 | 24.1 | 23.0 | >40 |
| 4 | >40 | 35.9 | 17.4 | >40 | >40 |
| 5 | 15.2 | 18.6 | 21.2 | 17.1 | 12.8 |
| 6 | 17.6 | 18.6 | 17.2 | 21.4 | 30.1 |
| 7 | 16.0 | 29.3 | 23.1 | 16.9 | 11.8 |
| 8 | 18.4 | >40 | 30.6 | 21.6 | 34.2 |
| DDP a | 3.1 | 10.2 | 9.1 | 17.5 | 12.0 |
Preliminary analysis of the structure–activity relationship revealed that the compounds with hydroxyl groups (3, 5–8) had greater cytotoxicity than the compounds without hydroxyl groups (2, 4). The reduction of ketone carbonyl group in 4 to hydroxyl group such as 5 or the introduction of 8-OH into 4 such as 3 led to a significant improvement of activity, except against A-549 cells, showing that 2-OH and 8-OH were beneficial for the improvement of activity against most of the cell lines, and the presence of 2-carbonyl group was able to increase activity against A-549 cells. However, the 2,8-dihydroxy compound 8 did not demonstrate higher activity than 2-hydroxy compound 5 or 8-hydroxy compound 3. The higher activity of 7 than 8 showed that the combination of 1,8-dihydroxy groups led to higher activity than that of 2,8-dihydroxy groups. In addition, the 1,8-dihydroxy compound 7 demonstrated higher activity than the corresponding 1-acetoxy-8-hydroxy compound 6 against HL-60, MCF-7 and SW480 but lower activity against SMMC-7721 and A-549.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}