Supraspliceosomes at Defined Functional States Portray the Pre-Assembled Nature of the Pre-mRNA Processing Machine in the Cell Nucleus

Abstract
:1. Introduction
2. Results and Discussion
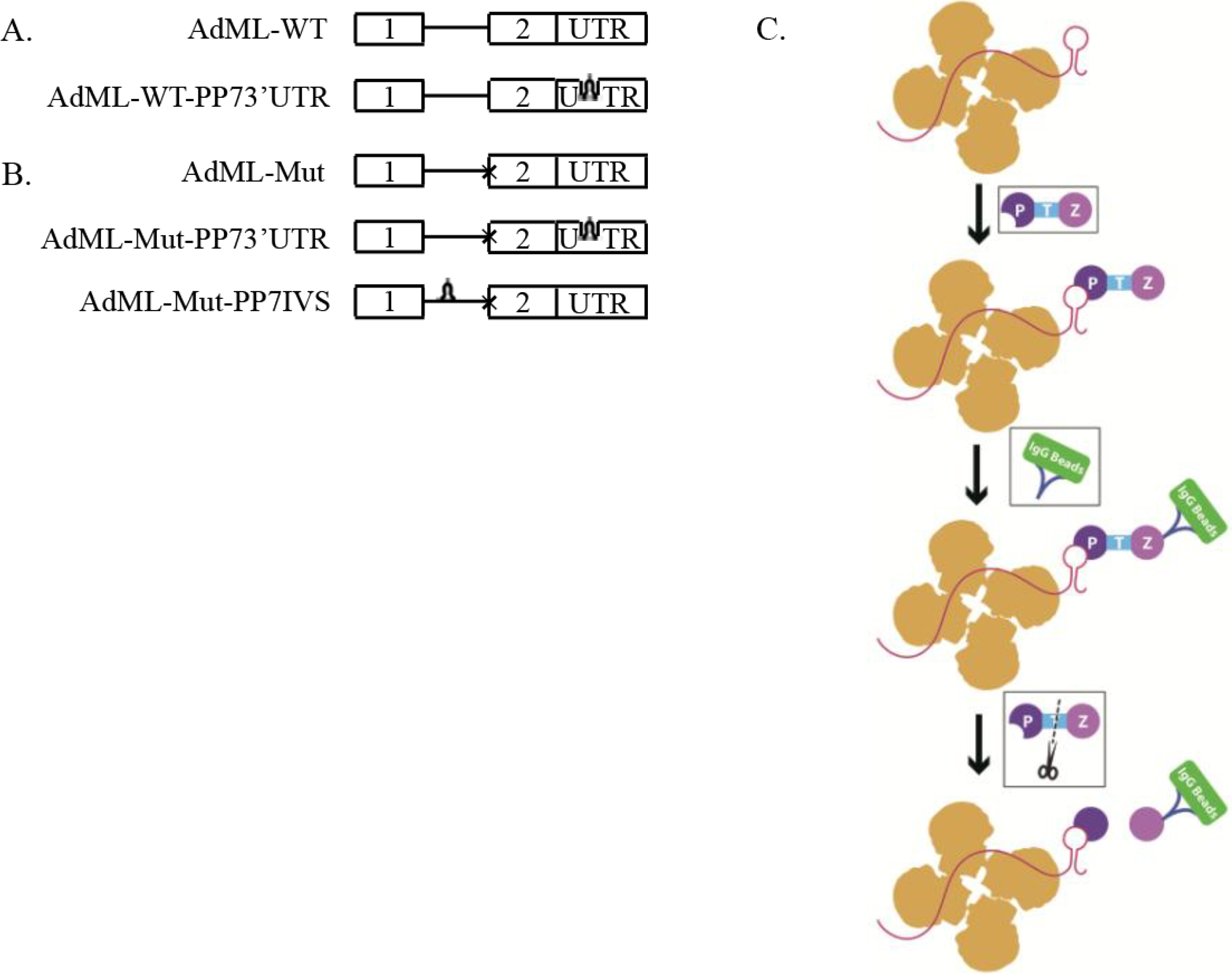
2.1. Generation of Stable Cell Lines Expressing Pseudomonas aeruginosa Phage 7 (PP7)-Tagged Adenovirus Major Late (AdML) Transcripts

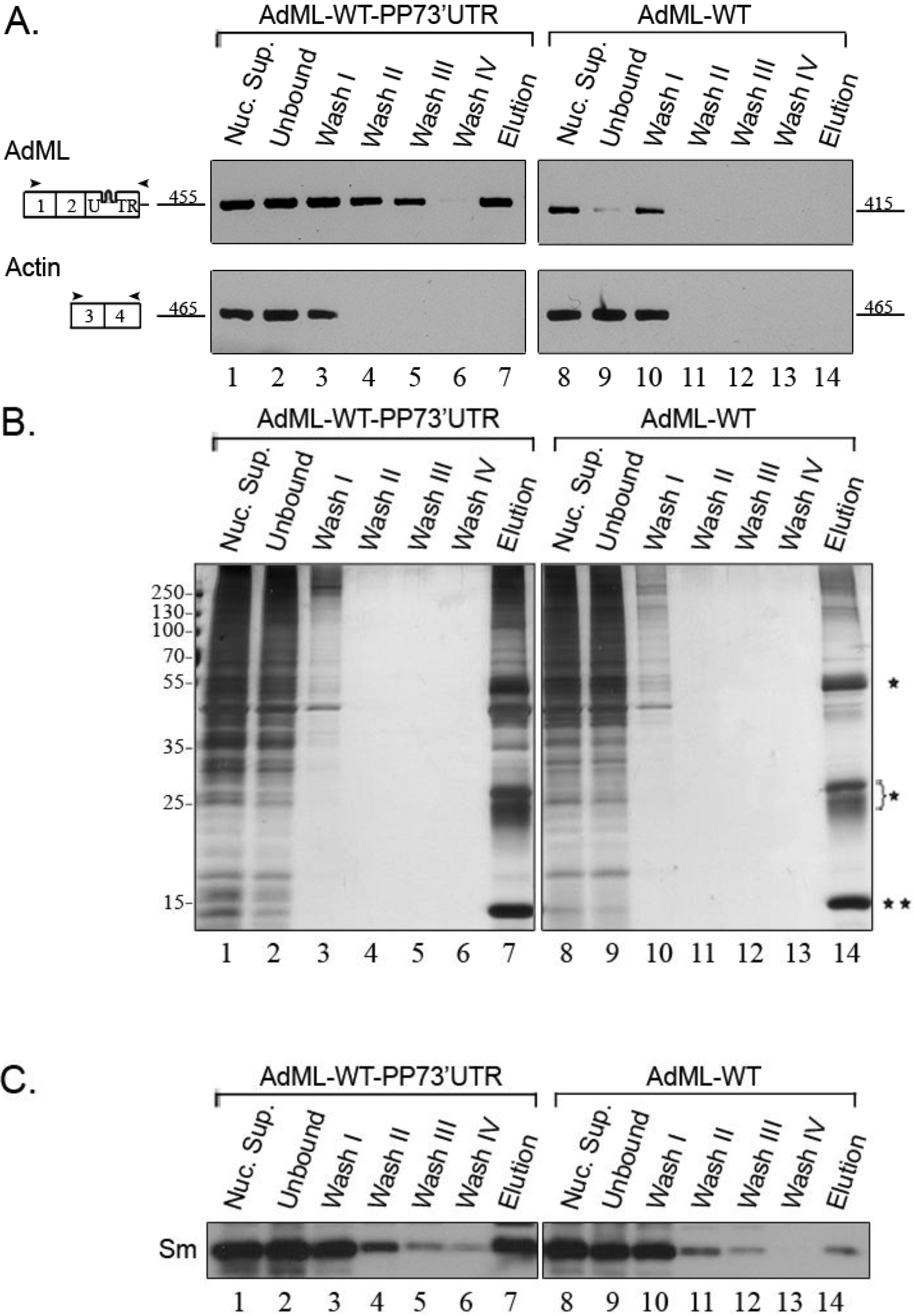
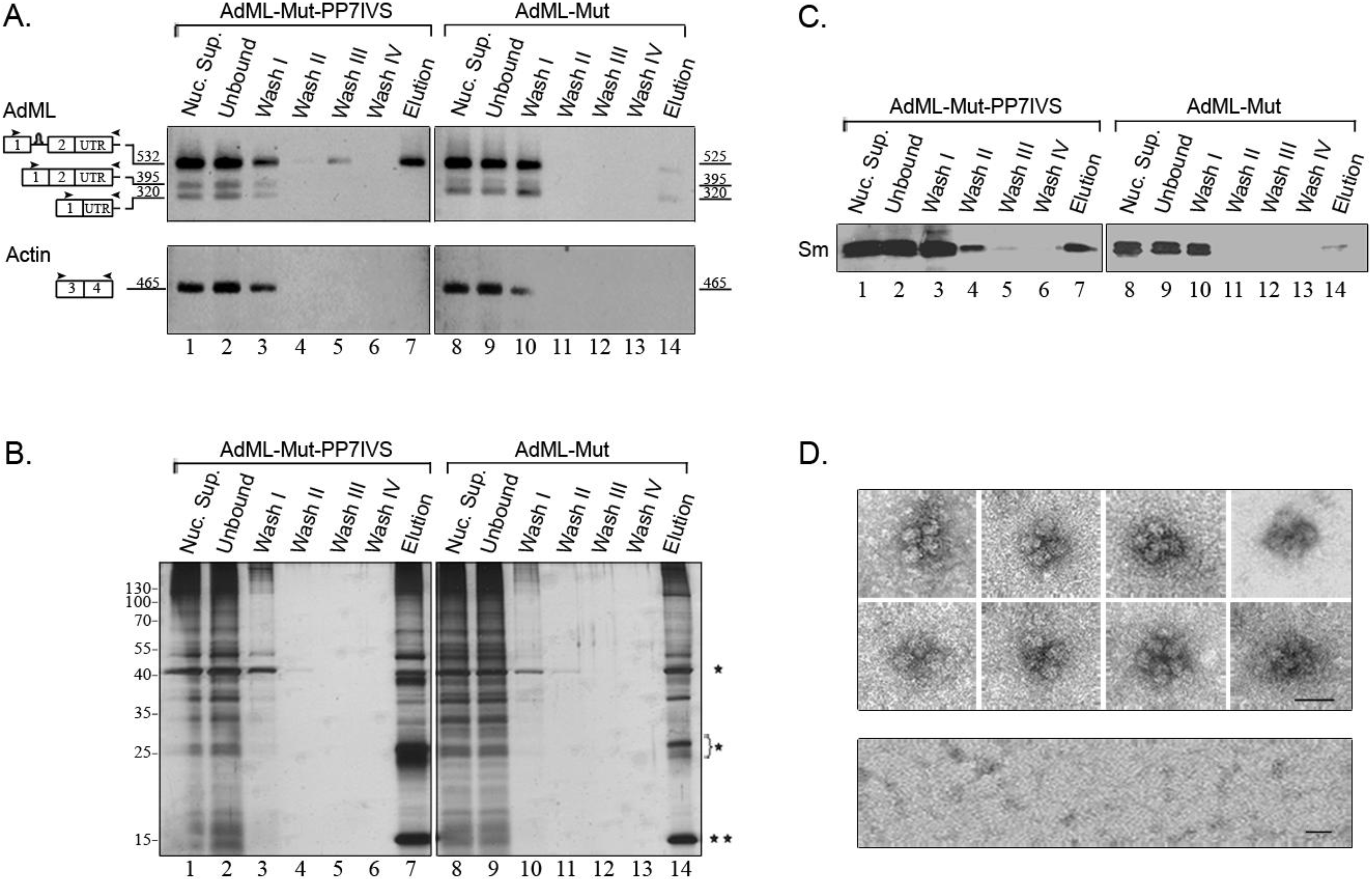
2.2. Affinity Purification of Complexes Assembled on AdML-WT Transcripts
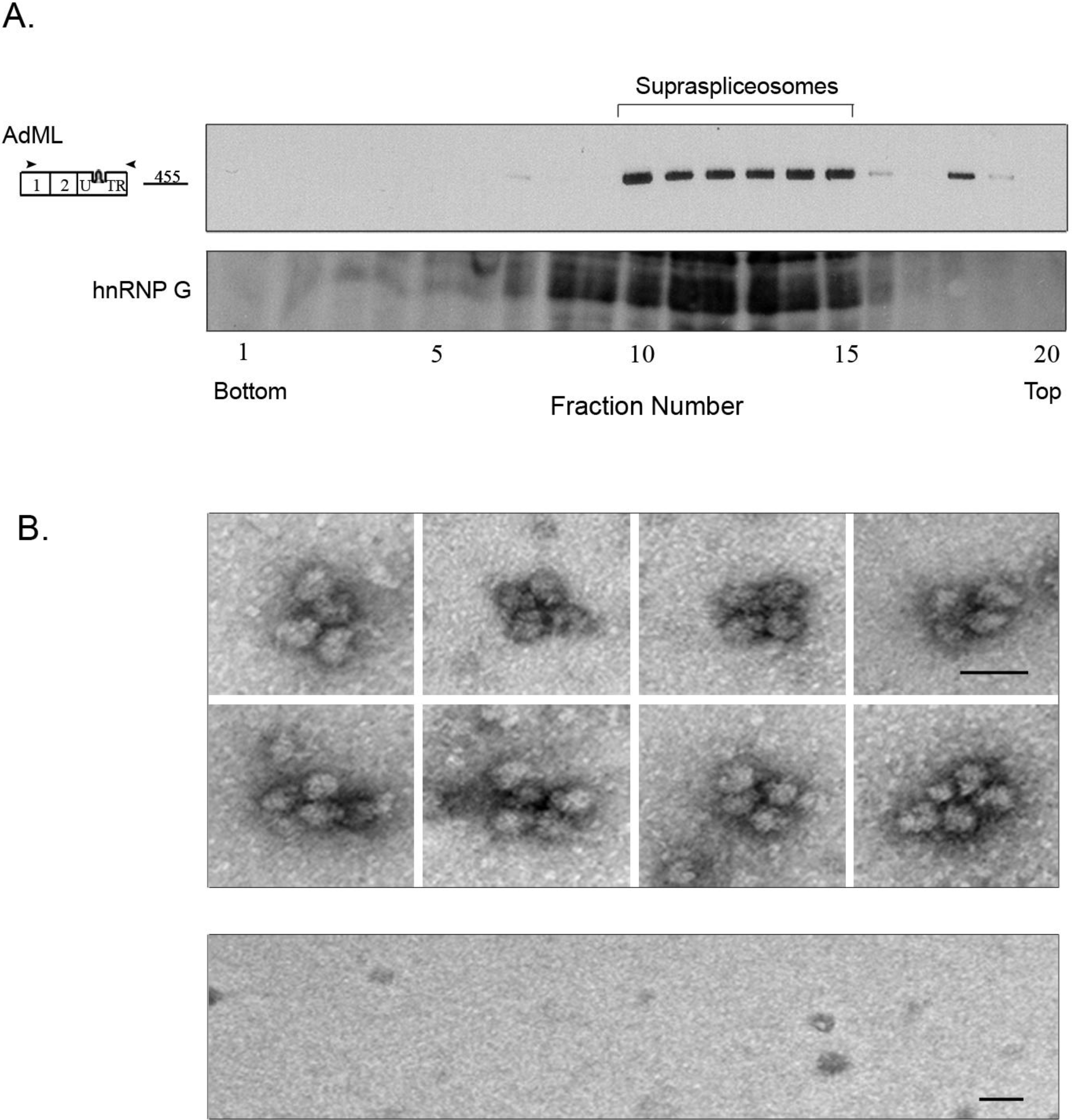
2.3. Affinity Purified PP7-Tagged AdML-WT Transcripts Are Assembled in Supraspliceosomes


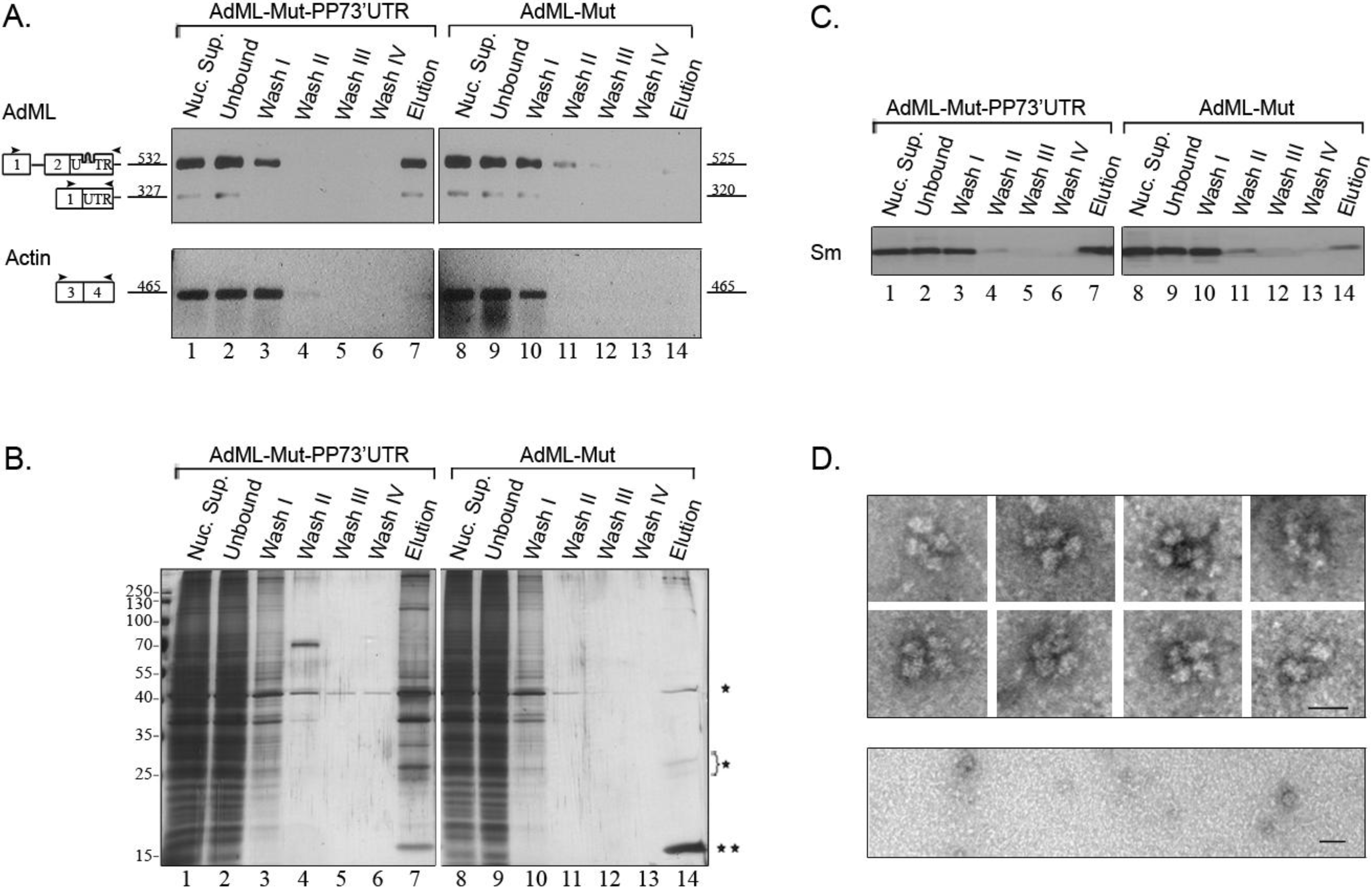
2.4. Affinity Purification of Supraspliceosomes Assembled on AdML-Mut Transcripts


2.5. Supraspliceosomes Isolated from Cell Nuclei Are Assembled with All Five Spliceosomal U snRNPs during the Steps of the Splicing Reaction

2.6. Protein Analysis of Affinity Purified Supraspliceosomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Protein | kDa | AdML-WT | AdML-Mut |
|---|---|---|---|---|
| U1 snRNP | ||||
| SNRNP70 | U1-70 k | 51.5 | + | |
| U2 snRNP | ||||
| SNRP A1 | U2A' | 28.4 | + | |
| SNRP B2 | U2B'' | 25.5 | + | |
| SF3A1 | SF3a120 | 88.9 | + | |
| SF3A2 | SF3a66 | 49.3 | + | |
| SF3A3 | SF3a60 | 58.8 | + | + |
| SF3B1 | SF3b155 | 145.8 | + | + |
| SF3B2 | SF3b145 | 100.2 | + | + |
| SF3B3 | SF3b130 | 135.6 | + | + |
| DDX46 | PRP5 | 117.4 | + | + |
| PHF5A | SF3b14b | 12.4 | + | + |
| U2 snRNP related | ||||
| U2AF2 | U2AF65 | 53.5 | + | + |
| U4/U6.U5 snRNP | ||||
| PRPF8 | U5-220k | 273.6 | + | + |
| SNRNP200 | U5-200k | 244.5 | + | + |
| EFTUD2 | U5-116k | 109.5 | + | + |
| PRPF6 | U5-102k | 106.9 | + | |
| DDX23 | U5-100k | 93.2 | + | + |
| PRPF3 | U4/U6-90k | 77.5 | + | |
| PRPF4 | U4/U6-60k | 58.4 | + | + |
| PRPF31 | U4/U6.U5-61k | 55.5 | + | |
| Sm proteins | ||||
| SNRP B | Sm B/B' | 24.6 | + | + |
| SNRP D1 | Sm D1 | 13.3 | + | + |
| SNRP D2 | Sm D2 | 13.5 | + | |
| SNRP D3 | Sm D3 | 13.9 | + | + |
| SNRP E | Sm E | 10.8 | + | + |
| CK005235 | Sm G Like | 85.4 | + | + |
| hPRP19/CDC5L complex | ||||
| PRPF19 | PRPF19 | 55.2 | + | + |
| CDC5L | CDC5 | 92.3 | + | |
| HSPA8 | HSP71 | 70.9 | + | + |
| PLRG1 | PRP45 | 57.2 | + | |
| XAB2 | SYF1 | 99.7 | + | |
| AQR | Aquarius | 156.8 | + | |
| Splicing factors | ||||
| DHX15 | DHX15 | 89.5 | + | + |
| DDX39 | DDX39 | 49.1 | + | + |
| DDX5 | DDX5 p68 | 69.1 | + | + |
| FUS | FUS TLS | 53.4 | + | + |
| SFPQ | hPOMp100 | 68.7 | + | + |
| TIA1 | p40-TIA-1 | 31.6 | + | |
| Splicing factors | ||||
| NONO | p54(nrb) | 54.3 | + | + |
| KHSRP | p75 | 73.1 | + | |
| PABPC1 | PABP1 | 70.7 | + | + |
| PUF60 | PUF60 | 59.9 | + | |
| RBM4 | RBM4 | 40.2 | + | |
| TARDBP | TDP43 | 31.8 | + | |
| DDX39B | UAP56 | 50.7 | + | + |
| YBX1 | YBX1 | 35.9 | + | + |
| hnRNP proteins | ||||
| HNRNPA0 | hnRNP A0 | 30.8 | + | + |
| HNRNPA1 | hnRNP A1 | 38.8 | + | + |
| HNRNPA3 | hnRNP A3 | 39.6 | + | + |
| HNRNPA2B1 | hnRNP A2/B1 | 37.4 | + | + |
| HNRNPC | hnRNP C1/C2 | 33.7 | + | + |
| RALY | hnRNP CL2 | 24.7 | + | + |
| HNRNPD | hnRNP D | 38.4 | + | |
| HNRNPDL | hnRNP DL | 46.4 | + | + |
| PCBP1 | hnRNP E1 | 37.5 | + | + |
| PCBP2 | hnRNP E2 | 33.9 | + | + |
| HNRNPF | hnRNP F | 45.7 | + | + |
| RBMX | hnRNP G | 42.3 | + | + |
| HNRNPH1 | hnRNP H1 | 49.2 | + | + |
| HNRNPH2 | hnRNP H2 | 49.3 | + | + |
| HNRNPH3 | hnRNP H3 | 36.9 | + | + |
| PTBP1 | hnRNP I | 57.2 | + | + |
| HNRNPK | hnRNP K | 51 | + | + |
| HNRNPL | hnRNP L | 64.1 | + | + |
| HNRNPM | hnRNP M | 77.5 | + | + |
| SYNCRIP | hnRNP Q | 69.6 | + | + |
| HNRNPR | hnRNP R | 70.9 | + | + |
| HNRNPU | hnRNP U | 90.6 | + | + |
| HNRNPUL1 | hnRNP UL1 | 95.7 | + | + |
| HNRNPUL2 | hnRNP UL2 | 85.1 | + | + |
| SR proteins | ||||
| SRSF1 | SF2/ASF | 28.3 | + | + |
| SFR3 | SRp20 | 19.3 | + | + |
| SFR10 | SFRS10 | 33.7 | + | + |
| SRRM2 | SRm300 | 299.6 | + | |
| SFRS11 | SRp54 | 24.8 | + | |
| SRSF2 | SRSF2 | 21.4 | + | |
| SRSF6 | SRp55 | 39.4 | + | + |
| SRSF7 | SRSF7 | 18.8 | + | + |
| 3' polyA/processing factors | ||||
| NUD21 | CPSF 25 | 26.2 | + | + |
| 3' polyA/processing factors | ||||
| CPSF7 | CPSF 59 | 51.1 | + | + |
| CPSF6 | CPSF 68 | 52.3 | + | + |
| CPSF3 | CPSF 73 | 73.5 | + | |
| CPSF2 | CPSF 100 | 88.5 | + | |
| CPSF1 | CPSF 160 | 152 | + | |
| CSTF2 | CSTF64 | 46.7 | + | |
| PABPN1 | PABII | 11 | + | + |
| SYMPK | Symplekin | 141.1 | + | |
| Cap binding | ||||
| NCBP1 | CBP80 | 91.8 | + | |
| mRNA export and surveillance | ||||
| UPF1 | UPF1 | 124.3 | + | + |
| ALYREF | ALYREF | 27.6 | + | |
| MAGOHB | MAGOHB | 17.3 | + | |
| EIF4A3 | eIF-4A-III | 46.9 | + | + |
| IGF2BP1 | IMP-1 | 63.5 | + | |
| RAN | RAN | 24.4 | + | + |
| XPO1 | Exportin | 123.4 | + | |
| RANBP2 | RANBP2 | 358.2 | + | + |
| RANGAP1 | RANGAP1 | 63.5 | + | |
| DDX3X | DDX3X | 73.2 | + | + |
| RNA metabolism | ||||
| CSL4 | CSL4 | 18.8 | + | |
| DDX1 | DBP-RB | 82.4 | + | + |
| DDX21 | DDX21 | 87.3 | + | + |
| DIS3 | DIS3 | 109.1 | + | |
| ELAVL1 | HuR | 39 | + | + |
| LRPPRC | LRP 130 | 157.9 | + | |
| ILF2 | NF45 | 43.1 | + | + |
| NF90 | NF90 | 76 | + | + |
| ILF3 | NF110 | 95.3 | + | |
| NCL | Nucleolin | 76.6 | + | + |
| RNA metabolism | ||||
| NPM1 | Nucleophosmin | 32.6 | + | + |
| PABPC4 | PABP-4 | 70.8 | + | + |
| EXOSC2 | RRP4 | 32.8 | + | |
| SKIV2L2 | SKIV2L2 | 117.8 | + | |
| SRRT | SRRT | 100.7 | + | + |
| TIAL1 | TIAL1 | 41.6 | + | |
| Accession | Name | kDa | AdML-WT-PP73'UTR | AdML-WT-PP73'UTR | AdML-WT-PP73'UTR | AdML-Mut-PP73'UTR | AdML-Mut-PP7int |
|---|---|---|---|---|---|---|---|
| P38152 | hnRNP G | 42.3 | 8 | 11 | 4 | 9 | |
| Q00839 | hnRNP U | 90.6 | 50 | 57 | 11 | ||
| B4DLR3 | hnRNP U | 86.9 | 18 | 31 | |||
| PO9651 | hnRNP A1 | 38.8 | 7 | ||||
| Q61PF2 | hnRNP A1 | 34.2 | 20 | 10 | 12 |

3. Experimental Section
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar]
- Burge, C.B.; Tuschl, T.H.; Sharp, P.A. The RNA World, 2nd ed.; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 525–560. [Google Scholar]
- Brow, D.A. Allosteric cascade of spliceosome activation. Annu. Rev. Genet. 2002, 36, 333–360. [Google Scholar]
- Will, C.L.; Lührmann, R. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 2001, 13, 290–301. [Google Scholar]
- Staley, J.P.; Guthrie, C. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell 1998, 92, 315–326. [Google Scholar] [CrossRef]
- Lin, S.; Fu, X.D. SR proteins and related factors in alternative splicing. Adv. Exp. Med. Biol. 2007, 623, 107–122. [Google Scholar]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef]
- Han, S.P.; Tang, Y.H.; Smith, R. Functional diversity of the hnRNPs: Past, present and perspectives. Biochem. J. 2010, 430, 379–392. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ryder, U.; Lamond, A.I.; Mann, M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002, 12, 1231–1245. [Google Scholar] [CrossRef]
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Luhrmann, R.; Stelzl, U. Dynamic protein–protein interaction wiring of the human spliceosome. Mol. Cell 2012, 45, 567–580. [Google Scholar] [CrossRef]
- Chen, Y.I.; Moore, R.E.; Ge, H.Y.; Young, M.K.; Lee, T.D.; Stevens, S.W. Proteomic analysis of in vivo-assembled pre-mRNA splicing complexes expands the catalog of participating factors. Nucleic Acids Res. 2007, 35, 3928–3944. [Google Scholar] [CrossRef]
- Sperling, J.; Azubel, M.; Sperling, R. Structure and function of the pre-mRNA splicing machine. Structure 2008, 16, 1605–1615. [Google Scholar] [CrossRef]
- Miriami, E.; Angenitzki, M.; Sperling, R.; Sperling, J. Magnesium cations are required for the association of U small nuclear ribonucleoproteins and SR proteins with pre-mRNA in 200S large nuclear ribonucleoprotein particles. J. Mol. Biol. 1995, 246, 254–263. [Google Scholar] [CrossRef]
- Azubel, M.; Habib, N.; Sperling, J.; Sperling, R. Native spliceosomes assemble with pre-mRNA to form supraspliceosomes. J. Mol. Biol. 2006, 356, 955–966. [Google Scholar] [CrossRef]
- Yitzhaki, S.; Miriami, E.; Sperling, J.; Sperling, R. Phosphorylated Ser/Arg-rich proteins: Limiting factors in the assembly of 200S large nuclear ribonucleoprotein particles. Proc. Natl. Acad. Sci. USA 1996, 93, 8830–8835. [Google Scholar] [CrossRef]
- Heinrich, B.; Zhang, Z.; Raitskin, O.; Hiller, M.; Benderska, N.; Hartmann, A.M.; Bracco, L.; Elliott, D.; Ben-Ari, S.; Soreq, H.; et al. Heterogeneous nuclear ribonucleoprotein G regulates splice site selection by binding to CC(A/C)-rich regions in pre-mRNA. J. Biol. Chem. 2009, 284, 14303–14315. [Google Scholar] [CrossRef]
- Markus, M.A.; Heinrich, B.; Raitskin, O.; Adams, D.J.; Mangs, H.; Goy, C.; Ladomery, M.; Sperling, R.; Stamm, S.; Morris, B.J. WT1 interacts with the splicing protein RBM4 and regulates its ability to modulate alternative splicing in vivo. Exp. Cell Res. 2006, 312, 3379–3388. [Google Scholar] [CrossRef]
- Yang, Y.-H.J.; Markus, A.M.; Mangs, H.A.; Raitskin, O.; Sperling, R.; Morris, B.J. ZRANB2 localizes to supraspliceosomes and influences the alternative splicing of multiple genes in the transcriptome. Mol. Biol. Rep. 2013, 40, 5381–5395. [Google Scholar] [CrossRef]
- Raitskin, O.; Angenitzki, M.; Sperling, J.; Sperling, R. Large nuclear RNP particles—The nuclear pre-mRNA processing machine. J. Struct. Biol. 2002, 140, 123–130. [Google Scholar] [CrossRef]
- Raitskin, O.; Cho, D.S.; Sperling, J.; Nishikura, K.; Sperling, R. RNA editing activity is associated with splicing factors in lnRNP particles: The nuclear pre-mRNA processing machinery. Proc. Natl. Acad. Sci. USA 2001, 98, 6571–6576. [Google Scholar] [CrossRef]
- Sperling, R.; Koster, A.J.; Melamed-Bessudo, C.; Rubinstein, A.; Angenitzki, M.; Berkovitch-Yellin, Z.; Sperling, J. Three-dimensional image reconstruction of large nuclear RNP (lnRNP) particles by automated electron tomography. J. Mol. Biol. 1997, 267, 570–583. [Google Scholar] [CrossRef]
- Medalia, O.; Typke, D.; Hegerl, R.; Angenitzki, M.; Sperling, J.; Sperling, R. Cryoelectron microscopy and cryoelectron tomography of the nuclear pre-mRNA processing machine. J. Struct. Biol. 2002, 138, 74–84. [Google Scholar] [CrossRef]
- Müller, S.; Wolpensinger, B.; Angenitzki, M.; Engel, A.; Sperling, J.; Sperling, R. A supraspliceosome model for large nuclear ribonucleoprotein particles based on mass determinations by scanning transmission electron microscopy. J. Mol. Biol. 1998, 283, 383–394. [Google Scholar] [CrossRef]
- Azubel, M.; Wolf, S.G.; Sperling, J.; Sperling, R. Three-dimensional structure of the native spliceosome by cryo-electron microscopy. Mol. Cell 2004, 15, 833–839. [Google Scholar] [CrossRef]
- Cohen-Krausz, S.; Sperling, R.; Sperling, J. Exploring the architecture of the intact supraspliceosome using electron microscopy. J. Mol. Biol. 2007, 368, 319–327. [Google Scholar]
- Spann, P.; Feinerman, M.; Sperling, J.; Sperling, R. Isolation and visualization of large compact ribonucleoprotein particles of specific nuclear RNAs. Proc. Natl. Acad. Sci. USA 1989, 86, 466–470. [Google Scholar] [CrossRef]
- Nilsen, T.W. RNA Structure and Function; Simons, R.W., Grunberg-Manago, M., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1998; pp. 279–307. [Google Scholar]
- Hogg, J.R.; Collins, K. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 2007, 13, 868–880. [Google Scholar] [CrossRef]
- Carey, J.; Uhlenbeck, O.C. Kinetic and thermodynamic characterization of the R17 coat protein-ribonucleic acid interaction. Biochemistry 1983, 22, 2610–2615. [Google Scholar] [CrossRef]
- Lim, F.; Downey, T.P.; Peabody, D.S. Translational repression and specific RNA binding by the coat protein of the Pseudomonas phage PP7. J. Biol. Chem. 2001, 276, 22507–22513. [Google Scholar]
- Sebbag-Sznajder, N.; Raitskin, O.; Angenitzki, M.; Sato, T.A.; Sperling, J.; Sperling, R. Regulation of alternative splicing within the supraspliceosome. J. Struct. Biol. 2012, 177, 152–159. [Google Scholar] [CrossRef]
- Baserga, S.J.; Steitz, J.A. The RNA World; Gesteland, R.F., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; pp. 359–381. [Google Scholar]
- Stevens, S.W.; Ryan, D.E.; Ge, H.Y.; Moore, R.E.; Young, M.K.; Lee, T.D.; Abelson, J. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol. Cell 2002, 9, 31–44. [Google Scholar]
- Iborra, F.J.; Jackson, D.A.; Cook, P.R. The path of transcripts from extra-nucleolar synthetic sites to nuclear pores: Transcripts in transit are concentrated in discrete structures containing SR proteins. J. Cell Sci. 1998, 111, 2269–2282. [Google Scholar]
- Schmidt, C.; Gronborg, M.; Deckert, J.; Bessonov, S.; Conrad, T.; Luhrmann, R.; Urlaub, H. Mass spectrometry-based relative quantification of proteins in precatalytic and catalytically active spliceosomes by metabolic labeling (SILAC), chemical labeling (iTRAQ), and label-free spectral count. RNA 2014, 20, 406–420. [Google Scholar]
- Nilsen, T.W. The spliceosome: No assembly required? Mol. Cell 2002, 9, 8–9. [Google Scholar] [CrossRef]
- Deckert, J.; Hartmuth, M.; Boehringer, D.; Behzadnia, N.; Will, C.L.; Kastner, B.; Stark, H.; Urlaub, H.; Luhrmann, R. Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol. Cell. Biol. 2006, 26, 5528–5543. [Google Scholar] [CrossRef]
- Boehringer, D.; Makarov, E.M.; Sander, B.; Makarova, O.V.; Kastner, B.; Luhrmann, R.; Stark, H. Three-dimensional structure of a pre-catalytic human spliceosomal complex B. Nat. Struct. Mol. Biol. 2004, 11, 463–468. [Google Scholar] [CrossRef]
- Rino, J.; Carmo-Fonseca, M. The spliceosome: A self-organized macromolecular machine in the nucleus? Trends Cell Biol. 2009, 19, 375–384. [Google Scholar] [CrossRef]
- Will, C.L.; Behrens, S.E.; Lührmann, R. Protein composition of mammalian spliceosomal snRNPs. Mol. Biol. Rep. 1993, 18, 121–126. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Chiu, Y.F.; Liu, Y.C.; Chiang, T.W.; Yeh, T.C.; Tseng, C.K.; Wu, N.Y.; Cheng, S.C. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Mol. Cell. Biol. 2009, 29, 5671–5678. [Google Scholar] [CrossRef]
- Liu, Y.C.; Chen, H.C.; Wu, N.Y.; Cheng, S.C. A novel splicing factor, Yju2, is associated with NTC and acts after Prp2 in promoting the first catalytic reaction of pre-mRNA splicing. Mol. Cell. Biol. 2007, 27, 5403–5413. [Google Scholar] [CrossRef]
- Warkocki, Z.; Odenwalder, P.; Schmitzova, J.; Platzmann, F.; Stark, H.; Urlaub, H.; Ficner, R.; Fabrizio, P.; Luhrmann, R. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat. Struct. Mol. Biol. 2009, 16, 1237–1243. [Google Scholar] [CrossRef]
- Villa, T.; Guthrie, C. The Isy1p component of the NineTeen complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev. 2005, 19, 1894–1904. [Google Scholar] [CrossRef]
- Fabrizio, P.; Dannenberg, J.; Dube, P.; Kastner, B.; Stark, H.; Urlaub, H.; Luhrmann, R. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol. Cell 2009, 36, 593–608. [Google Scholar]
- Kolev, N.G.; Yario, T.A.; Benson, E.; Steitz, J.A. Conserved motifs in both CPSF73 and CPSF100 are required to assemble the active endonuclease for histone mRNA 3' end maturation. EMBO Rep. 2008, 9, 1013–1018. [Google Scholar] [CrossRef]
- Mandel, C.R.; Kaneko, S.; Zhang, H.; Gebauer, D.; Vethantham, V.; Manley, J.L.; Tong, L. Polyadenylation factor CPSF-73 is the pre-mRNA 3' end-processing endonuclease. Nature 2006, 444, 953–956. [Google Scholar] [CrossRef]
- Mandel, C.R.; Bai, Y.; Tong, L. Protein factors in pre-mRNA 3' end processing. Cell. Mol. Life Sci. 2008, 65, 1099–1122. [Google Scholar] [CrossRef]
- Agranat, L.; Raitskin, O.; Sperling, J.; Sperling, R. The editing enzyme ADAR1 and the mRNA surveillance protein hUpf1 interact in the cell nucleus. Proc. Natl. Acad. Sci. USA 2008, 105, 5028–5033. [Google Scholar] [CrossRef]
- Taniguchi, I.; Ohno, M. ATP-dependent recruitment of export factor Aly/REF onto intronless mRNAs by RNA helicase UAP56. Mol. Cell. Biol. 2008, 28, 601–608. [Google Scholar]
- Sperling, R.; Sperling, J. RNP Particles, Splicing and Autoimmune Diseases; Schenkel, J., Ed.; Springer: Heidelberg, Germany, 1998; pp. 29–47. [Google Scholar]
- Lerner, E.A.; Lerner, M.R.; Janeway, L.A.; Steitz, J.A. Monoclonal antibodies to nucleic acid containing cellular constituents: Probes for molecular biology and autoimmune disease. Proc. Natl. Acad. Sci. USA 1981, 78, 2737–2741. [Google Scholar]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar]
- Ishihama, Y.; Rappsilber, J.; Andersen, J.S.; Mann, M. Microcolumns with self-assembled particle frits for proteomics. J. Chromatogr. A 2002, 979, 233–239. [Google Scholar] [CrossRef]
- Uniprot. Available online: http://www.uniprot.org/ (accessed on 9 January 2013).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kotzer-Nevo, H.; De Lima Alves, F.; Rappsilber, J.; Sperling, J.; Sperling, R. Supraspliceosomes at Defined Functional States Portray the Pre-Assembled Nature of the Pre-mRNA Processing Machine in the Cell Nucleus. Int. J. Mol. Sci. 2014, 15, 11637-11664. https://doi.org/10.3390/ijms150711637
Kotzer-Nevo H, De Lima Alves F, Rappsilber J, Sperling J, Sperling R. Supraspliceosomes at Defined Functional States Portray the Pre-Assembled Nature of the Pre-mRNA Processing Machine in the Cell Nucleus. International Journal of Molecular Sciences. 2014; 15(7):11637-11664. https://doi.org/10.3390/ijms150711637
Chicago/Turabian StyleKotzer-Nevo, Hani, Flavia De Lima Alves, Juri Rappsilber, Joseph Sperling, and Ruth Sperling. 2014. "Supraspliceosomes at Defined Functional States Portray the Pre-Assembled Nature of the Pre-mRNA Processing Machine in the Cell Nucleus" International Journal of Molecular Sciences 15, no. 7: 11637-11664. https://doi.org/10.3390/ijms150711637
APA StyleKotzer-Nevo, H., De Lima Alves, F., Rappsilber, J., Sperling, J., & Sperling, R. (2014). Supraspliceosomes at Defined Functional States Portray the Pre-Assembled Nature of the Pre-mRNA Processing Machine in the Cell Nucleus. International Journal of Molecular Sciences, 15(7), 11637-11664. https://doi.org/10.3390/ijms150711637