1. Introduction
MicroRNAs (miRNA) are an extensive series of small non-coding RNAs (18–25 nt) with an increasingly recognizable importance in the control of profound regulation of many biological processes in cell proliferation, apoptosis, organogenesis, hematopoiesis, tumorgenesis by regulating gene expression [
1]. They are transcribed from intergenic or intronic sequences as long precursors termed pri-miRNAs that are sequentially processed by the enzyme Drosha into ~70 nt pre-miRNAs and Dicer into approximate 22-nt mature miRNAs in the cytoplasm [
2,
3]. Mature miRNAs regulate gene expression post-transcriptionally by binding principally to the 3'-untranslated regions (3'-UTR) of specific mRNA, thereby guiding the effector proteins of RNAi-induced silencing complex (RISC) into close proximity with the mRNAs [
4,
5], then the RISC complex will either cleave the target messenger or inhibit its translation [
6].
Identification of miRNA–mRNA interaction has been a great challenge. Bioinformatics algorithms have been developed to predict putative miRNA–mRNA interaction [
7,
8,
9]. It has been found that seven to eight nt at the 5' end of the miRNA is the most important determinant for target specificity which called “seed sequence” [
10,
11]. Many computational approaches for the prediction of miRNA–mRNA interaction are based on complete base pairing to the target mRNA at seed region of miRNA. Although some target genes have been successfully predicted for a few miRNAs, a large number of potential targets are falsely predicted because of perfect seed pairing may not necessarily be a reliable predictor for miRNA interactions [
12]. Therefore, biochemical approaches have been designed to address this issue.
Recently, some biochemical assays are applied to identify new mRNA targets for miRNAs based on affinity purification of labeled miRNA–mRNA complex [
13,
14,
15,
16,
17]. However, only a few experiment methods have been used to find new miRNAs that regulate one certain gene [
18,
19]. Here, we have developed a more convenient experimental approach using biotinylated DNA oligonucleotides mix to search for miRNAs that regulate specific mRNA which is fused to enhanced green fluorescent protein (
EGFP) mRNA (
Figure 1), in which the complex of miRNA/mRNA was purified by streptavidin beads through binding to the biotinylated DNA oligonucleotides mix. With the fusion
EGFP tag, we just need to design biotinylated DNA antisense capture oligonucletides specific to the
EGFP mRNA instead of designing different antisense oligonucletides for various genes and investigate the transfection efficiency by fluorescence. We selected the programmed cell death protein 4 (
PDCD4) and miR-21 which are well studied in miRNA–mRNA interaction for this study [
20,
21,
22]. The 3'-UTR of
PDCD4 mRNA contains a MRE (miRNA response element) of miR-21 and was cloned into the downstream of
EGFP. Using the binding between miRNA and mRNA which was stabilized by formaldehyde and the affinity interaction between biotinylated DNA and streptavidin bead (Thermo Scientific, Waltham, MA, USA), miRNAs targeting the 3'-UTR of
PDCD4 were successfully isolated. As a known target miRNA of
PDCD4, miR-21 was successfully detected in the isolated miRNAs with polymerase chain reaction (PCR). This experimental approach provides us a simple assay to directly seek out the target miRNAs for any interested gene. Furthermore, this approach is suitable for any miRNAs that target anywhere within the full length of mRNA transcript.
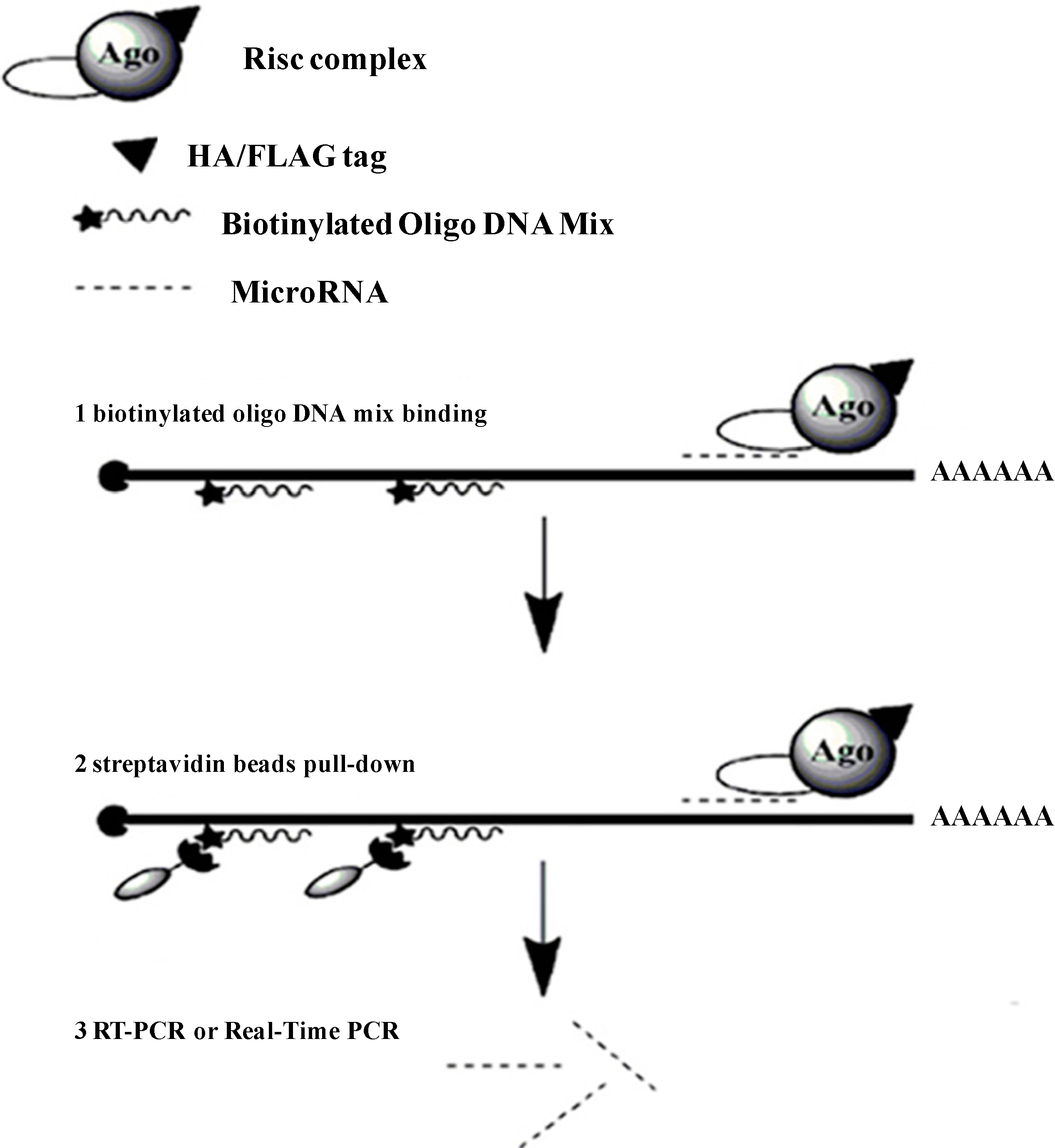
Figure 1.
The pull-down strategy of the determination of the microRNAs (miRNAs) that target to a specific gene. Extracts from the cells that over-expressed a tagged Argonaute protein are first incubated with biotinylated oligo DNA mix, and then affinity purified on streptavidin beads. MiRNAs are quantified by real time quantitative PCR (qRT-PCR).
Figure 1.
The pull-down strategy of the determination of the microRNAs (miRNAs) that target to a specific gene. Extracts from the cells that over-expressed a tagged Argonaute protein are first incubated with biotinylated oligo DNA mix, and then affinity purified on streptavidin beads. MiRNAs are quantified by real time quantitative PCR (qRT-PCR).
2. Results and Discussion
As reports,
PDCD4 is regulated by miR-21 via its binding to the specific site within 3'-UTR in HeLa cells [
20,
21]. Therefore, we chose
PDCD4 and miR-21 as a model for this study. About 500 nt 3'-UTR of
PDCD4 which includes miR-21 binding site was inserted into the downstream of
EGFP (
Figure 2A) and 3'-UTR of
β-ACTIN was also inserted as a negative control which is not regulated by miR-21 [
21]. Meanwhile, a stop codon was added between
EGFP and 3'-UTR. As a result, we can fusion any interested gene into the downstream of
EGFP that does not influence the
EGFP expression. Expression of
EGFP-PDCD4 3'-UTR and
EGFP-ACTIN 3'-UTR chimeric mRNA were detected by fluorescence microscope. The results show that those chimeric mRNA could be transcripted and translated in HeLa cells (
Figure 2B). Then we investigated the binding of miR-21 to the chimeric mRNAs by quantitative real-time PCR using miR-21 mimics transfected with the plasmids above in HeLa cells, and the results showed us that chimeric mRNA of
EGFP-PDCD4 3'-UTR can be bound by miR-21 (
Figure 2C).
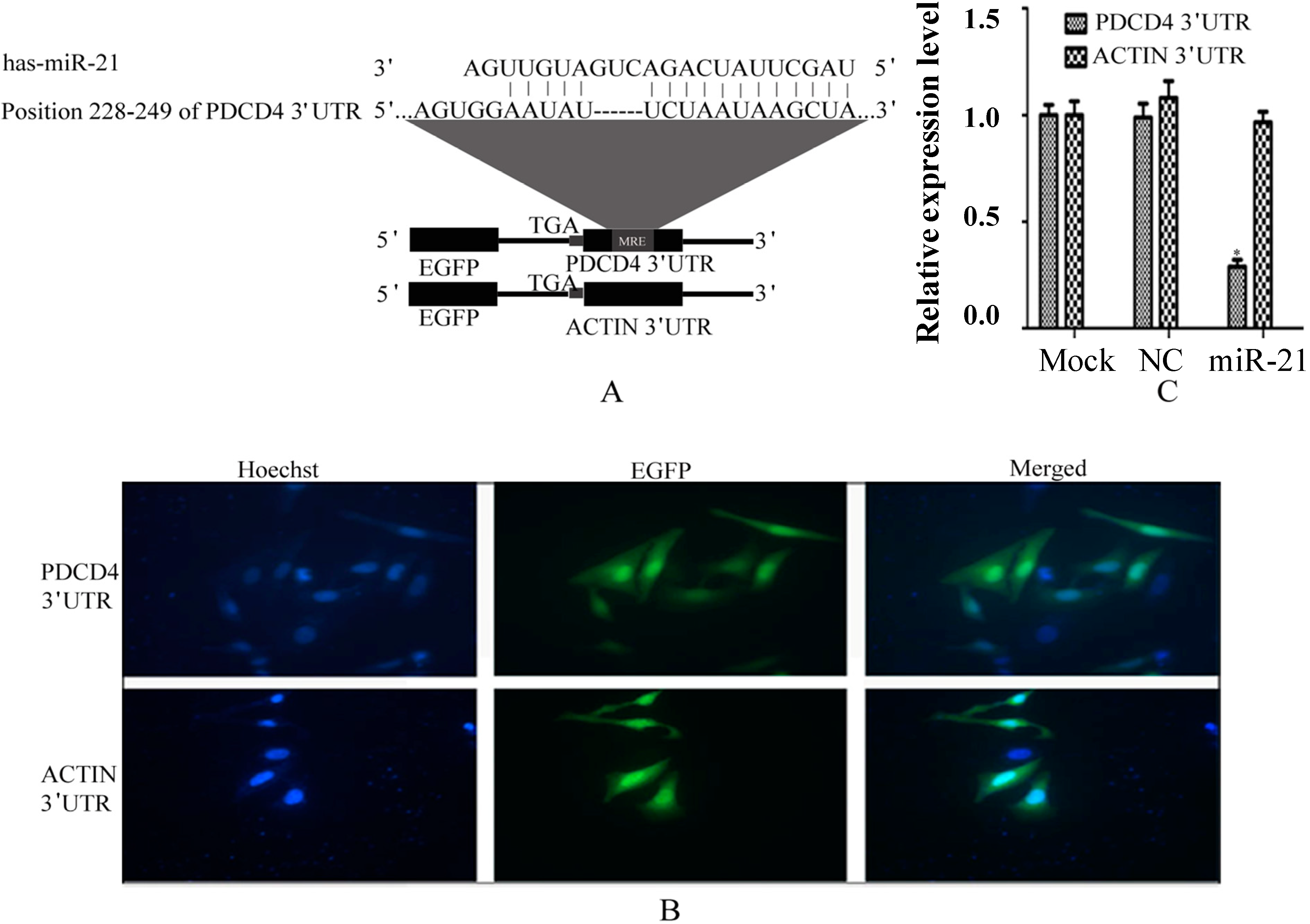
Figure 2.
The 3'-untranslated regions (3'-UTR) of programmed cell death protein 4 (PDCD4) was regulated by miR-21. (A) Diagram of reporter constructs with EGFP tag. The 3'-UTR of PDCD4 with predicted MRE of miR-21 or 3'-UTR of ACTIN without MRE were inserted into the downstream of EGFP gene of pCMV-enhanced green fluorescent protein (EGFP) vector. A stop code was added before the 3'-UTR; (B) The expression of EGFP-3'-UTR were detected by fluorescence microscope. Nuclear was stained by Hoechst 33258; and (C) Repression of EGFP-3'-UTR by the interaction between miR-21 and the 3'-UTR. Data are shown as the mean ± SD of three independent experiments.* p < 0.05.
Figure 2.
The 3'-untranslated regions (3'-UTR) of programmed cell death protein 4 (PDCD4) was regulated by miR-21. (A) Diagram of reporter constructs with EGFP tag. The 3'-UTR of PDCD4 with predicted MRE of miR-21 or 3'-UTR of ACTIN without MRE were inserted into the downstream of EGFP gene of pCMV-enhanced green fluorescent protein (EGFP) vector. A stop code was added before the 3'-UTR; (B) The expression of EGFP-3'-UTR were detected by fluorescence microscope. Nuclear was stained by Hoechst 33258; and (C) Repression of EGFP-3'-UTR by the interaction between miR-21 and the 3'-UTR. Data are shown as the mean ± SD of three independent experiments.* p < 0.05.
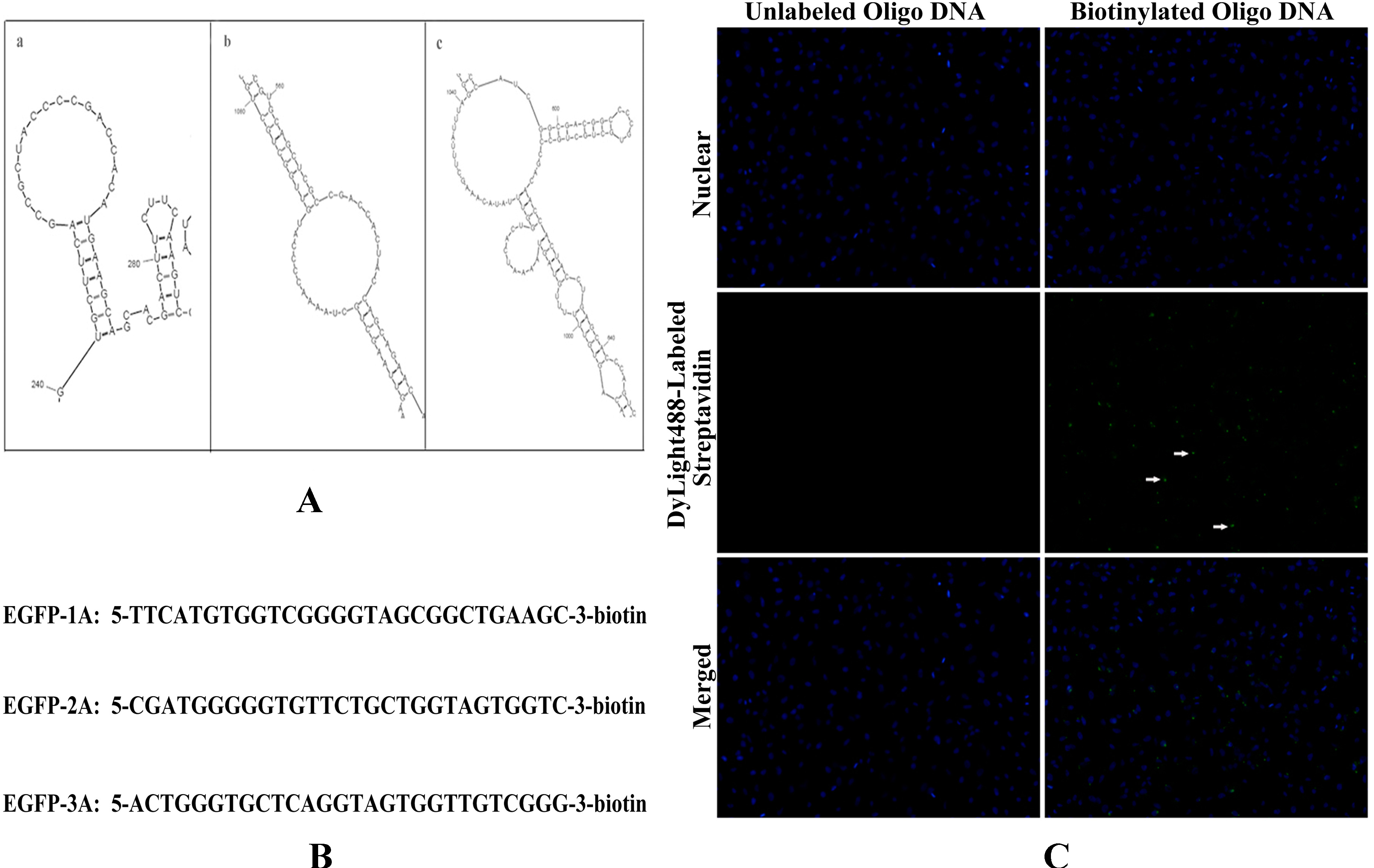
The secondary structures of the
EGFP mRNA were predicted by software M-Fold (mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form). Three exposed single-stranded regions which were present in the first three of the most thermodynamically stringent structures were selected to design DNA oligonucleotide capture sequences (
Figure 3A). According to the three predicted secondary structures of
EGFP mRNA, three 28-nt antisense oligo DNAs targeting to different single-stranded region were designed (
Figure 3B). The three antisense oligo DNAs were labeled with biotin at 3'- end and mixed to enhance the binding force to
EGFP mRNA. To verify the antisense oligo DNAs whether labeled with biotin, we utilized affinity between streptavidin and biotin. Streptavidin conjugated with green fluorescent moiety (Dylight 488-streptavidin) was added into HeLa cells which was transfected with biotinylated oligo DNAs and detected by fluorescence microscope. Compared with unlabeled antisense oligo DNAs, biotinylated oligo DNAs could interact with Dylight 488-streptavidin and show green point distribution in cytoplasm (show as white arrow,
Figure 3C). It is demonstrated that antisense oligo DNAs are well biotinylated.
Figure 3.
Antisense oligo DNAs of EGFP mRNA were labeled with biotin. (A) The secondary structures of the EGFP mRNA were predicted by software M-Fold; (B) The sequence of antisense oligo DNA targeting to EGFP mRNA were biotinylated at 3'-end; and (C) Biotinylated oligo DNA were detected by fluorescent microscope through Dylight 488-streptavidin.
Figure 3.
Antisense oligo DNAs of EGFP mRNA were labeled with biotin. (A) The secondary structures of the EGFP mRNA were predicted by software M-Fold; (B) The sequence of antisense oligo DNA targeting to EGFP mRNA were biotinylated at 3'-end; and (C) Biotinylated oligo DNA were detected by fluorescent microscope through Dylight 488-streptavidin.
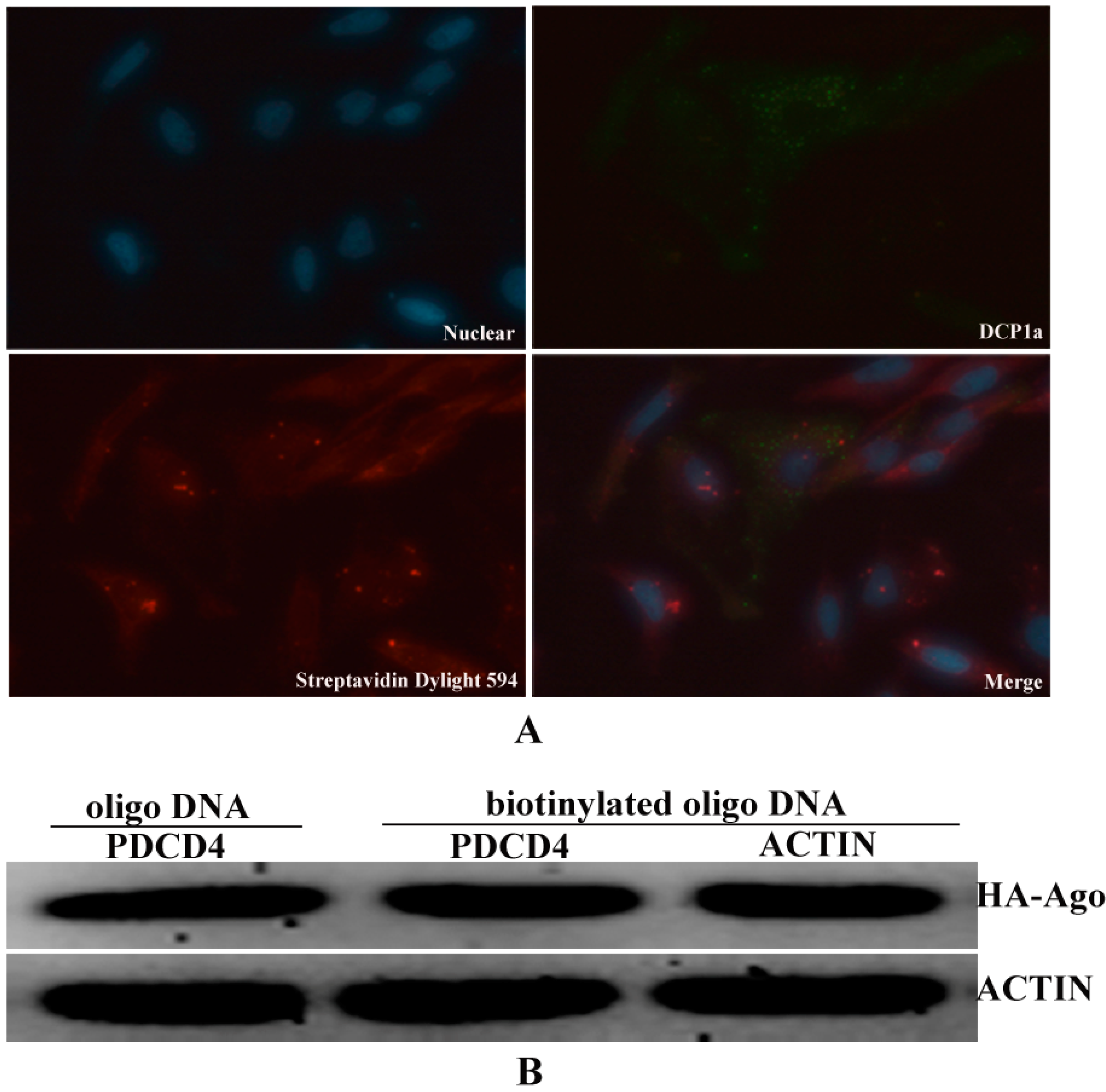
The biotinylated antisense oligo DNAs showed point distribution in the cytoplasm, so we speculated that the biotinylated oligo DNAs may combine with target
EGFP mRNA in some complexes that related to the processing and modification of mRNA, such as P-body which was also contributed to the regulation of miRNA and Dcp1a is a marker gene of P-body [
23]. To demonstrate the possible interaction between biotinylated oligo DNAs and P-body, the Immunoflurescent assays were performed as described above. The results revealed that biotinylated oligo DNAs and Dcp1a were not co-localized in the cytoplasm (
Figure 4A). It is proved that biotinylated oligo DNAs were not localized in P-body. As for this, the binding of biotinylated oligo DNAs to target
EGFP mRNA may not lead the transcriptional inhibition through the model of P-body [
24]. To increase the possible binding of miRNA to target mRNA, we overexpressed AGO2 protein which is a key component of RISC complex that regulated miRNA–mRNA interaction (
Figure 4B).
Figure 4.
Over-expression of Argonaute 2 protein is to increasing the bingding force between miRNA and mRNA. (A) Biotinylated oligo DNA were not localization with P-body in cells; and (B) Flag/HA-Ago 2 were detected in the extracts of cells that transfected with Flag/HA-Ago 2 vector.
Figure 4.
Over-expression of Argonaute 2 protein is to increasing the bingding force between miRNA and mRNA. (A) Biotinylated oligo DNA were not localization with P-body in cells; and (B) Flag/HA-Ago 2 were detected in the extracts of cells that transfected with Flag/HA-Ago 2 vector.
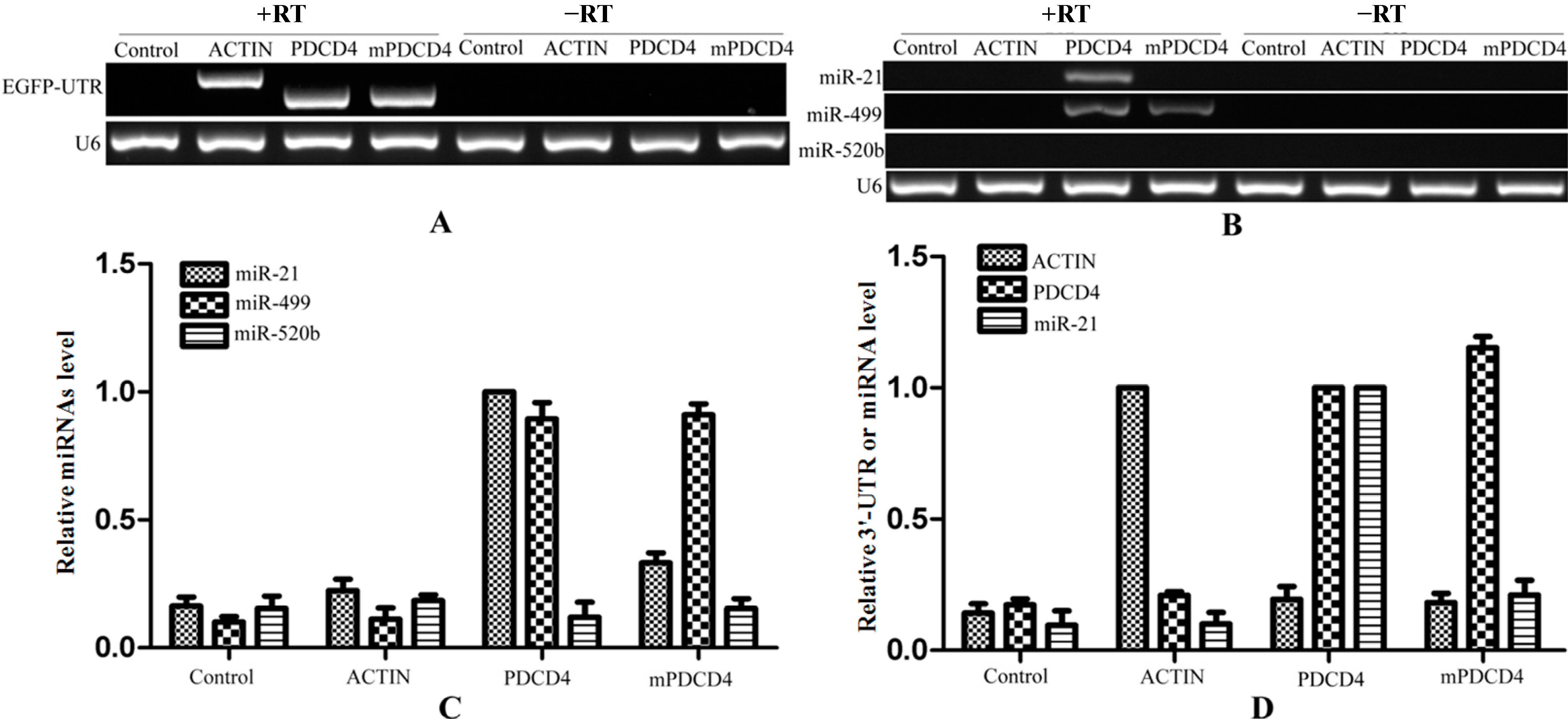
Biotinylated oligo DNAs can combine with target mRNA under the denatured condition and then enriched by streptavidin beads depending on biotin-streptavidin affinity interaction. After 24 h of the transfection of the plasmids in HeLa cells, the complexes of miRNA–mRNA–RBPs (RNA binding proteins) were pulled-down by biotinylated oligo DNAs that bound to target EGFP mRNA. During the process of pull-down, 37% formalin was firstly used for cross-linking among miRNA–mRNA–RBPs which increases the strength of interaction. The cross-link can be reversed under 65 °C for several hours [
25]. Hence, the cross-linked protein-RNA complex would not dissociate under 70 °C in a short period of time for unfolding the secondary structures of mRNAs that made the binding of biotinylated oligo DNAs to target
EGFP mRNA more convenient. Then, we used the streptavidin beads for affinity purification of biotinylated oligo DNAs combined with the miRNA–mRNA–RBPs complexes. Finally, the streptavidin beads were washed three times and the binding RNAs were extracted with Trizol (TaKaRa, Dalian, China) and reverse transcripted using the mRNA and miRNA first strand cDNA synthesis kit independently. During the pull-down process, the 3'-UTR of
PDCD4 that contains a mutated binding site of miR-21 was cloned as a control (mPDCD4). To verify that the target chimeric mRNAs were in the products of pull-down, we amplified the 3'-UTRs of
PDCD4,
mPDCD4 and
β-ACTIN using the synthesized cDNA as template (
Figure 5A). The forward primers were both designed within the sequence of
EGFP, while the reverse primers were in the 3'-UTR respectively. Therefore, the products would not be amplified from endogenous
PDCD4 or
β-ACTIN. As shown in the
Figure 5A,
PDCD4,
mPDCD4 and
β-ACTIN could be amplified from the templates that reverse transcripted with reverse transcriptase, while not from the templates without reverse transcriptase which excluded the pollution of plasmids transfected. It is demonstrated that the target genes of biotinylated oligo DNAs were pulled-down successfully. Then, we used semi-quantitative PCR and real time quantitative PCR (qRT-PCR) to prove that miR-21 is successfully pulled down followed with the target mRNAs. MicroRNA of
PDCD4 was contained in the products of streptavidin-biotin pull-down. However, the amount of miR-21 was very low, so KOD-FX DNA polymerase (Toyobo, Osaka, Japan) was used to amplify the target miRNA (
Figure 5B). MiR-21 was only detected in the product of streptavidin-biotin pull-down from the cells that were transfected with 3'-UTR-PDCD4, while not shown in the cells transfected with 3'-UTR-β-ACTIN or mutated 3'-UTR-PDCD4 (mPDCD4). The product that was pulled down from the cells that were transfected with 3'-UTR-PDCD4 by the oligo DNAs without biotin labeled was also taken as a negative control. All of the templates that were reversely transcribed without reverse transcriptase could not detect the existence of miR-21 (right lines in
Figure 5B), and U6 fragments which were added in the elution buffer equally were taken as an endogenous control. The same results were obtained from qRT-PCR (
Figure 5C). MiR-21 had a large amount of enrichment only in the product of streptavidin-biotin pull-down from the cells that were transfected with 3'-UTR-PDCD4, while had less in the cells transfected with 3'-UTR-β-ACTIN and the negative control.
Figure 5.
Validation of miRNAs targeting to single mRNA isolation technique. The RNA in the products of pull-down were reverse transcripted and as templates for amplifying. The chimeric mRNAs of EGFP-3'-UTR were detected from corresponding products of pull-down (A); The enrichment of miRNAs targeting to 3'-UTR-PDCD4 were shown in the pull-down products using semi-quantitative PCR (B) and qRT-PCR (C); The 3'-UTRs and miR-21 levels were detected from the pull-down products (D). RT: Reverse Transcriptase.
Figure 5.
Validation of miRNAs targeting to single mRNA isolation technique. The RNA in the products of pull-down were reverse transcripted and as templates for amplifying. The chimeric mRNAs of EGFP-3'-UTR were detected from corresponding products of pull-down (A); The enrichment of miRNAs targeting to 3'-UTR-PDCD4 were shown in the pull-down products using semi-quantitative PCR (B) and qRT-PCR (C); The 3'-UTRs and miR-21 levels were detected from the pull-down products (D). RT: Reverse Transcriptase.
MiR-499, as another target of
PDCD4 [
26,
27], was enriched in the product of streptavidin-biotin pull-down from the cells that were transfected with 3'-UTR-PDCD4 or mutated 3'-UTR-PDCD4, while miR-520b that do not target to 3'-UTR of
PDCD4 was not shown the enrichment (
Figure 5B,C). To verify the purification method, qRT-PCR was performed with 3'-UTRs and miR-21 primers. This verification was performed using the 2
−ΔΔCt method. As shown in
Figure 5D, both the 3'-UTRs of
ß-ACTIN and
PDCD4 were only enrichment in the product that was transfected with corresponding EGFP-3'-UTR plasmid. The levels of
ß-ACTIN and
PDCD4 in the product that was transfected with 3'-UTR of
ACTIN or
PDCD4 were artificially set to 1.0 respectively. The levels of miR-21 had a larger enrichment in the product transfected with 3'-UTR of
PDCD4 (artificially set to 1.0), while less in the product transfected with mutated 3'-UTR of
PDCD4 and other control products. These results demonstrated that our method for isolating the target miRNAs is efficient and specific.
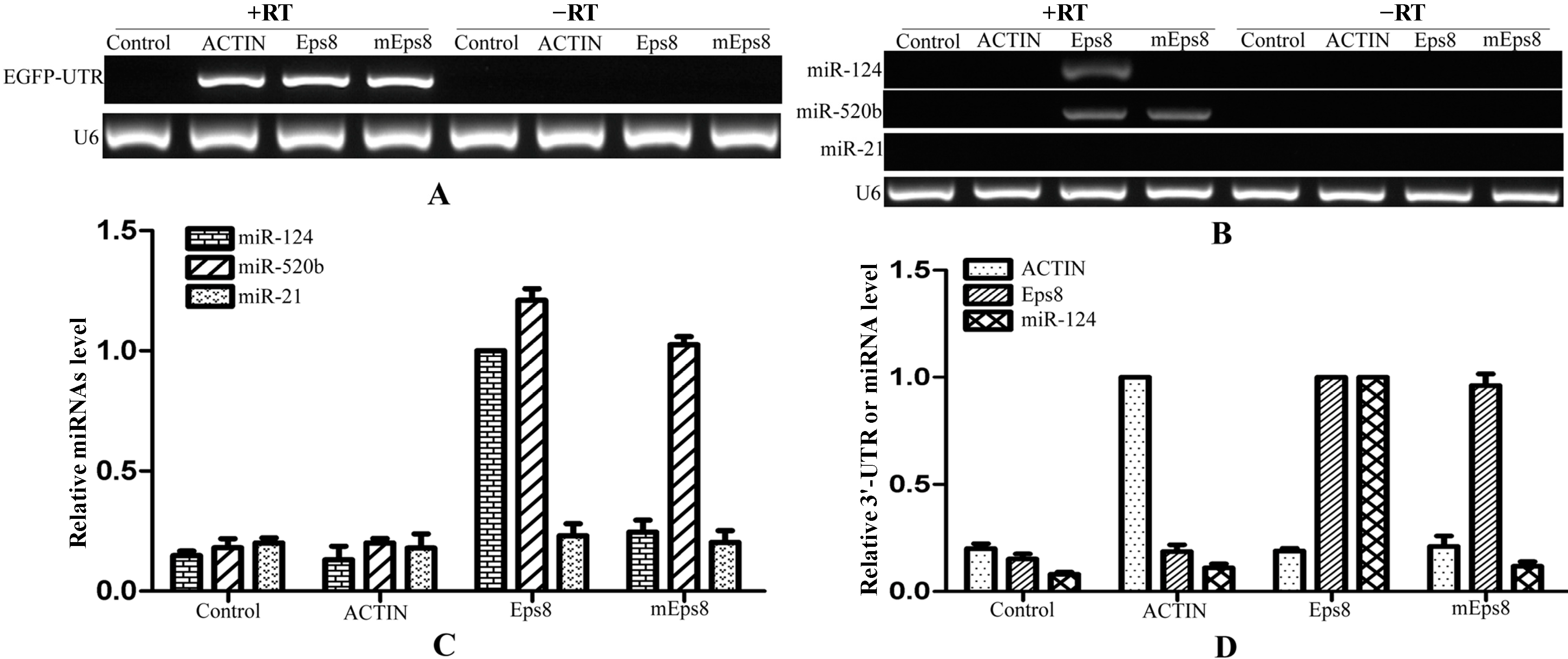
Figure 6.
mRNA:miRNAs isolation technique for Eps8 and its target miRNAs. (A) The enrichment of RNA chimeric mRNAs of EGFP-3'-UTR demonstrated the efficiency of isolation technique; (B) The regulated miRNAs of Eps8 were obtained from the product that was transfected with 3'-UTR-Eps8 through semi-quantitative PCR. At the same time, miRNAs that without binding sites could not be amplified from these products; (C) The levels of detected miRNAs were verified using qRT-PCR after the process of isolation technique; and (D) qRT-PCR showing the relationship between mRNA and their target miRNA after the isolation. RT: Reverse Transcriptase.
Figure 6.
mRNA:miRNAs isolation technique for Eps8 and its target miRNAs. (A) The enrichment of RNA chimeric mRNAs of EGFP-3'-UTR demonstrated the efficiency of isolation technique; (B) The regulated miRNAs of Eps8 were obtained from the product that was transfected with 3'-UTR-Eps8 through semi-quantitative PCR. At the same time, miRNAs that without binding sites could not be amplified from these products; (C) The levels of detected miRNAs were verified using qRT-PCR after the process of isolation technique; and (D) qRT-PCR showing the relationship between mRNA and their target miRNA after the isolation. RT: Reverse Transcriptase.
To validate our affinity purification procedure, another gene and its known regulated miRNAs were processed as described above. It had been reported that
Eps8 could be down-regulated by miR-124 and miR-520b through bind to the 3'-UTR [
28]. Over 500 nt 3'-UTR of
Eps8 that contains the binding sites of these two target miRNAs was inserted into the downstream of
EGFP. Another 3'-UTR of
Eps8 that contains a mutated binding site of miR-124 was also cloned into the downstream of
EGFP as a control. After pull-down, the three
EGFP-3'-UTRs were detected from the products that reversely transcripted with reverse transcriptase, while not from the products without reverse transcriptase (
Figure 6A). The target miRNAs of
Eps8 were detected using semi-quantified PCR (
Figure 6B). MiR-124 and miR-520b could be amplified from the product that transfected with the 3'-UTR of
Eps8, while miR-124 could not be detected from the product that transfected with mutated 3'-UTR-
Eps8. The same results were verified using qRT-PCR (
Figure 6C). MiR-124 showed a larger enrichment in the product that was transfected with 3'-UTR-
Eps8 (set to 1.0). The mutated 3'-UTR-
Eps8 had an obviously negative effect on the pull-down efficiency of miR-124, while no effect on miR-520b. Meanwhile, miR-21 that do not target to 3'-UTR of
Eps8 had no enrichment in any product (
Figure 6B,C). Finally, we also detected the enrichment of UTRs and miR-124 (
Figure 6D). These results clearly demonstrated a causal relationship between the enrichment of target miR-124 and the 3'-UTR-
Eps8, the same as miR-21 and 3'-UTR-
PDCD4 (
Figure 5D). It is suggested that our experimental approach can isolate the target miRNAs for a single gene successfully.
3. Experimental Methods
3.1. Cell Culture and Transfection
Human cervical cancer HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo, Waltham, MA, USA) containing 10% fetal bovine serum (Thermo, Waltham, MA, USA), 100 units/mL penicillin (Beyotim, Beijing, China), 100 µg/mL streptomycin (Beyotim, Beijing, China) and 2 mM l-glutamine (Invitrogen, Carlsbad, CA, USA) at 37 °C and 5% CO2. Antisense strand DNAs labeled with biotin were synthesized by Genepharma. Expression vectors described in this study were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
3.2. Plasmid Constructs
The 3'-UTR of
PDCD4,
Eps8 and
β-ACTIN were amplified from HeLa cDNA and then inserted into the 3'-end of the
EGFP gene of the expression vector pEGFP-C1 (Clontech, Palo Alto, CA, USA) between BglII and EcoRI sites. The primers used were:
PDCD4 3'-UTR-F (5'-AGGAGATCTTTTTGAGTACAAGGCATTTCTGAC-3') and
PDCD4 3'-UTR-R (5'-AAGGAATTCAGATGTTCCAGCCACCTTTTACTT-3'),
Eps8 3'-UTR-F (5'-ATGAGATCTCTATAGAGCATTCTCAAGAC-3') and
Eps8 3'-UTR-R (5'-AGGGAATTCCATTTATTAACAGACAGCAA-3'),
β-ACTIN 3'-UTR-F (5'-TTAAGATCTCCATAGTCCACCGCAAAT-3') and
β-ACTIN 3'-UTR-R (5'-ATAGAATTCCTGCCTCCACCCACTCCC-3'). Both of the forward primers were added a stop code TAG at the 3'-flanking of restriction sites. Translation of 3'-UTR was prevented by the stop code, but ineffective to the mRNA of
EGFP. The binding site of miRNA during 3'-UTR of
PDCD4 or
Eps8 was deleted mutated and cloned into downstream of
EGFP according to the references [
20,
28]. Finally, plasmids were sequenced and characterized.
3.4. Western Blot
HeLa cells were washed twice with ice-cold PBS and lysed in lysis buffer (1 M Tris–HCl, pH 7.5, 4 M NaCl, 1% tristonx-100, 10 mg/mL deoxycholate sodium, 1 mg/mL Sodium dodecyl sulfate, 1% cocktail). 20 μg of soluble protein was separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Darmstadt, Germany). Primary antibody against flag (Sigma, St. Louis, MO, USA) was used in this report. The protein was detected using a HRP-conjugated secondary antibody and SuperSignal West Pico Chemiluminescent substrate kits (Pierce, Wayland, MA, USA).
3.5. Immunocytochemical Staining
HeLa cells were grown on sterilized coverslips until 60%–80% confluent and then transfected using Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA) for 24 h. Cells were washed twice with ice-cold PBS and then fixed with 4% (w/v) paraformaldehyde in PBS for 10 min, and permeabilized with acetone and methanol (1:1) for 30 s. Cells were then incubated with primary antibodies diluted in PBS with 10% (v/v) normal goat serum for 1 h and with the secondary antibody under the same conditions. The primary antibody was anti-streptavidin (Thermo, Waltham, MA, USA) that marked by DyLight 488 (Molecular Probes, KPL, Gaithersburg, MD, USA). Hoechst 33258 (Sigma, St. Louis, MO, USA) was used to stain the nuclei. Fluorescence on the processed slips was analyzed using a fluorescence microscope (Zeiss, Oberkochen, Germany).
3.6. Streptavidin-Biotin Pull-Down
HeLa cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 24 h. 37% formalin was added for 10 min at room temperature and then additional 1 mL 10× Glycine for 5 min to stop the cross-linking reaction. The cells were washed twice with ice-cold PBS and collected into a new RNase-Free tube. HeLa cells were lysed with RIPA buffer (50 mM Tris–HCl (pH 7.2), 150 mM NaCl, 1% (v/v) Triton X-100, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS) in ice-water for 30 min. Centrifuged and the supernatant was denatured in 70 °C for 5 min to depolymerize the secondary structure. Supernatant was mixed with biotinylated DNA of EGFP for 1 h at room temperature and then incubated with equilibriumed streptavidin beads. Streptavidin beads were washed 4 times with 10% glycerol, 20 mM Tris (pH 8.0), 0.2 mM EDTA, 0.5% NP-40, 0.35 M KCl, 1 mM DTT. Finally, the complex of biotin-DNA:mRNA:miRNA with the magnetic beads were collected by centrifugation.
3.7. qRT-PCR
For qRT-PCR, RNAs in the complex of biotin-DNA:mRNA:miRNA were directly extracted using Trizol (Invitrogen, Carlsbad, CA, USA) after being incubated with Proteinase K (Sigma, St. Louis, MO, USA) in 55 °C for 10 min. The extracted RNAs were reverse transcripted with commercially available NCode™ miRNA First-Strand cDNA Synthesis and qRT-PCR assay (Promega, Madison, WI, USA). The extracted RNAs were also reverse transcripted with Reverse Transcription System (Promega, Madison, WI, USA) and used for the amplifying 3'-UTRs of PDCD4 and β-ACTIN. The resulting cDNA was amplified using oligonucleotide primers and SYBR Green MasterMix (TaKaRa, Dalian, China) for qRT-PCR. Relative copies of miR-21 were determined using the 2−ΔΔCt method and the U6 fragments served as an endogenous control. Oligonucleotide primers used were as follows: EGFP forward, 5-GCAGAAGAACGGCATCAAG-3; PDCD4 reverse, 5-ATGTTCCAGCCACCTTTTACTT-3; ACTIN reverse, 5-CTGCCTCCACCCACTCCC-3; Eps8 reverse, 5-TTAGAGAAATTATCTAGATTC-3. The forward qRT-PCR primers of the miRNAs were purchased from GeneCopoeia (Guangzhou, China). The reverse qRT-PCR primers of the miRNAs were used NCode™ miRNA First-Strand cDNA Synthesis and qRT-PCR kit (Invitrogen, Carlsbad, CA, USA).
3.8. Statical Analysis
GraphPad Prism 5 was used for statistical analysis. All results were presented as mean ± SD. The significance of differences between samples were performed by Student’s t-test and defined as *p < 0.05.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





