Deactivation of 6-Aminocoumarin Intramolecular Charge Transfer Excited State through Hydrogen Bonding

Abstract
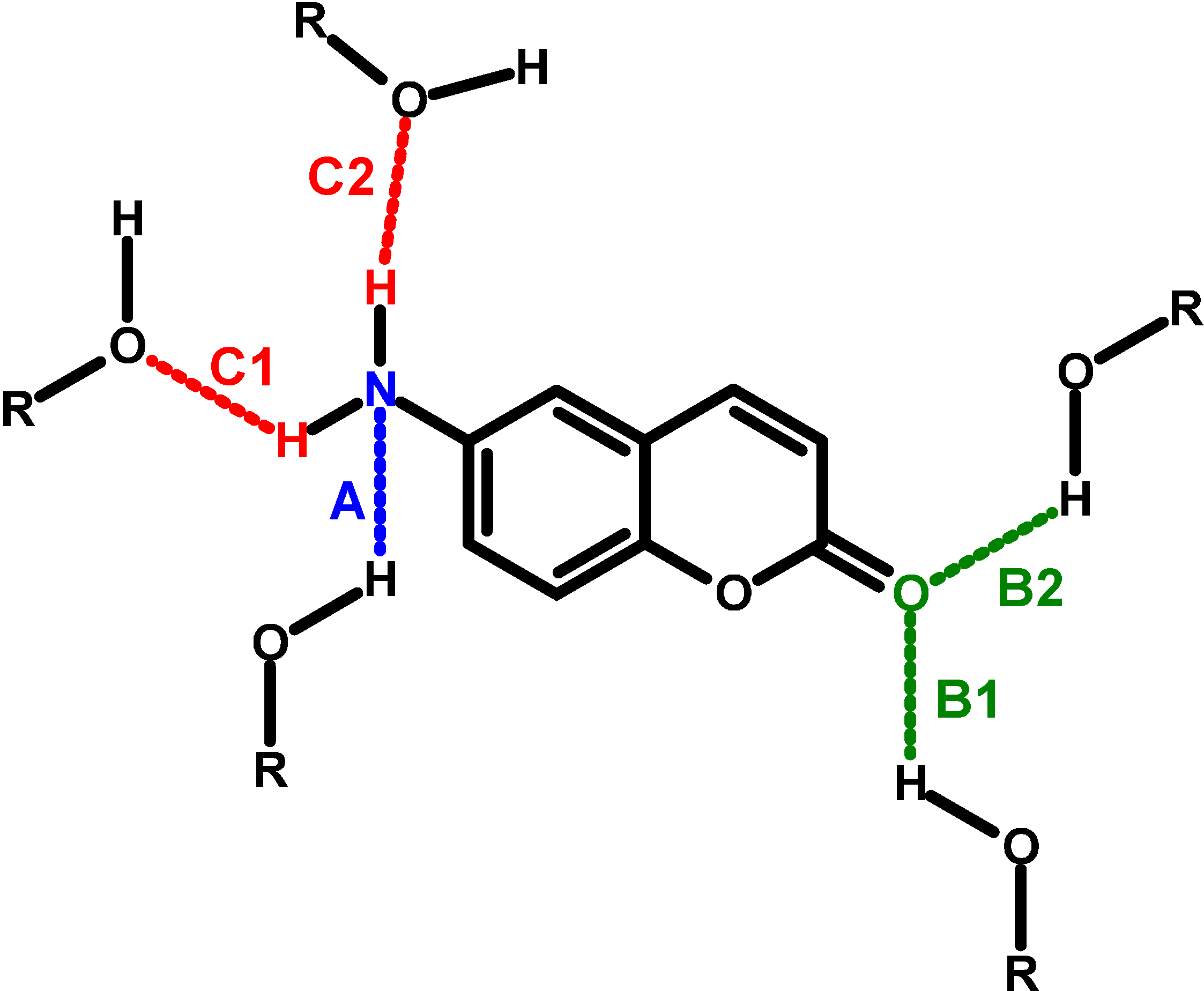
:1. Introduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent |  (cm−1) (cm−1) |  (cm−1) (cm−1) |  (cm−1) (cm−1) |  (cm−1) (cm−1) |  (cm−1) (cm−1) | ε( ) (mol−1·dm3·cm−1) | f(ε, n2) | ε a | n a | α a | β a |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1-Chloro-n-hexadecane b | 27,250 | 21,030 | 4780 | 4350 | 6220 | − | 0.110 | 3.70 | 1.450 | 0.00 | 0.00 |
| 1-Chloro-n-decane b | 27,250 | 20,750 | 4640 | 4370 | 6500 | 2860 | 0.145 | 4.58 | 1.438 | 0.00 | 0.00 |
| 1-Chloro-n-octane | 27,250 | 20,650 | 4600 | 4350 | 6600 | 3240 | 0.160 | 5.05 | 1.430 | 0.00 | 0.00 |
| 1-Chloro-n-hexane | 27,250 c | 20,490 | 4650 | 4320 | 6760 | 3270 | 0.184 | 6.10 | 1.419 | 0.00 | 0.00 |
| 1-Chloro-n-butane | 27,250 c | 20,400 | 4650 | 4300 | 6850 | 3350 | 0.209 | 7.39 | 1.400 | 0.00 | 0.00 |
| 1-Chloro-n-propane b | 27,250 | 20,300 | 4600 | 4400 | 6950 | 3450 | 0.226 | 8.59 | 1.386 | 0.00 | 0.00 |
| Acrylonitrile | 26,950 | 18,470 | 4660 | 4270 | 8480 | 2850 | 0.287 | 33.00 | 1.388 | 0.00 | 0.25 |
| Propionitrile b | 26,880 | 18,810 | 4750 | 4340 | 8070 | 2750 | 0.292 | 28.26 | 1.363 | 0.00 | 0.37 |
| DBE | 26,950 | 20,070 | 4620 | 4330 | 6880 | 2820 | 0.096 | 3.08 | 1.397 | 0.00 | 0.46 |
| THF | 26,670 | 19,430 | 4840 | 4480 | 7240 | 2930 | 0.210 | 7.58 | 1.405 | 0.00 | 0.55 |
| DMF | 26,100 | 17,850 | 4810 | 4220 | 8250 | 2800 | 0.275 | 36.71 | 1.428 | 0.00 | 0.69 |
| DMSO | 25,910 c | 17,500 | 4800 | 4180 | 8410 | 2860 | 0.264 | 46.45 | 1.477 | 0.00 | 0.76 |
| HMPA | 25,250 | 17,550 | 5200 | 4170 | 7700 | 2340 | 0.261 | 29.30 | 1.457 | 0.00 | 1.00 |
), molar extinction coefficient; n, refraction coefficient; ε, dielectric constant; α, Kamlet-Taft’s solvatochromic parameter related to hydrogen-bond donating ability; β, Kamlet-Taft’s solvatochromic parameter related to hydrogen-bond accepting ability; f(ε, n2) = (ε − 1)/(2ε + 1) – (n2 − 1)/(2n2 + 1); a From reference [13]; b From reference [4]. A typing error in for 6AC in 1-chloro-n-propane in references [4,5] was noticed; c From reference [3]; DBE, di-n-butyl ether; HMPA, hexamethylphosphoramide; THF, tetrahydrofuran; DMSO, dimethyl sulphoxide; and DMF, N,N-dimethylformamide.2. Results and Discussion
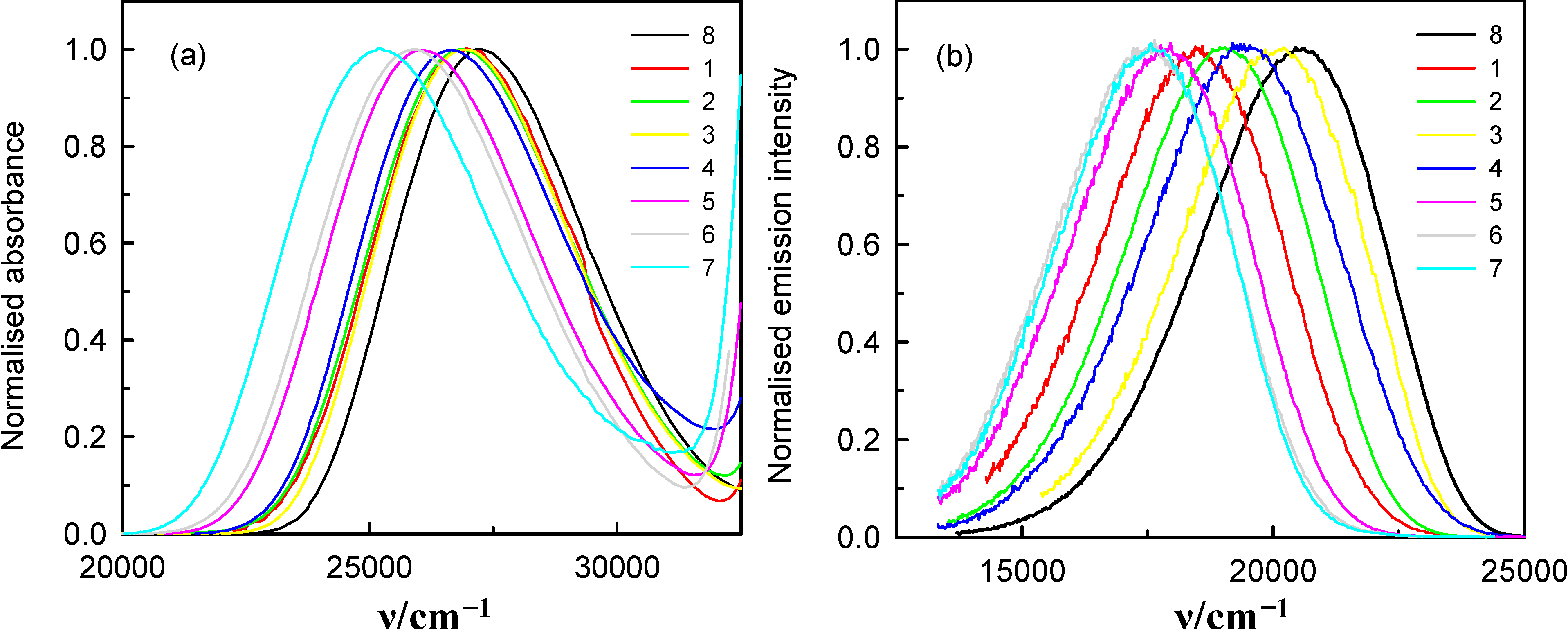
2.1. Spectral Properties of 6AC in Aprotic Hydrogen-Bond Forming Solvents
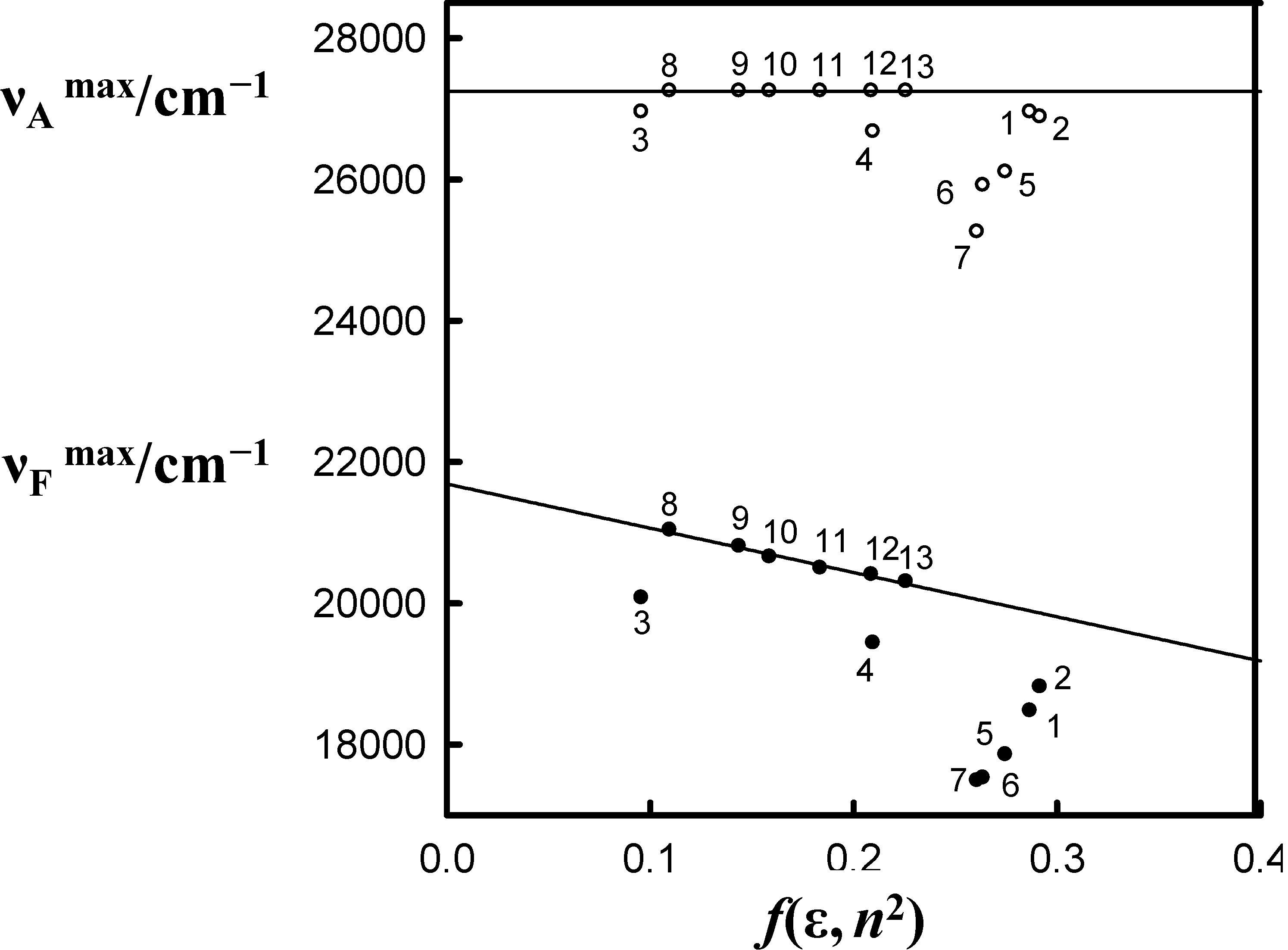
 , in the spectra of 6AC in all solvents, which can form hydrogen bonds of acceptor character only, shift towards longer wavelength with respect to that in the spectrum of 6AC in 1-chloro-n-propane. The shapes of the long-wavelength band (Figure 2a) do not differ significantly in the spectra taken for all solvents used, but FWHM (full width at half maximum) in the absorption spectra, , increases slightly, while the batochromic shift increases significantly with increasing β Kamlet-Taft’s solvent parameter (Table 1). Similarly to all in the fluorescence spectra of 6AC in aprotic hydrogen-bond forming solvents (see Figure 2b and Table 1) are shifted towards longer wavelength with respect to that in the spectrum of 6AC in 1-chloro-n-propane. The shape of the fluorescence band (Figure 2b) and the FWHM value of the fluorescence spectra of 6AC, , are practically the same in all solvents used, similarly as that of the absorption spectra, but is lower than that for 6AC in 1-chloro-n-propane. No influence of the excitation wavelength, λexc, on the position and shape of the fluorescence spectrum was noted, similarly as reported earlier in references [4,5,39]. Very large Stokes shifts, in comparison with those of 7-aminocoumarins (e.g., [40,41]), have been observed between the absorption and fluorescence maxima (as reported earlier in reference [39]). This indicates that the changes in energy of solute–solvent specific (hydrogen bond formation) and non-specific interactions have a great influence on the static spectroscopic properties of 6AC. Table 1 lists the absorption and fluorescence maxima ( and , respectively), FWHM values of the absorption and fluorescence ( and , respectively) and the Stokes shifts, = − , of 6AC in different solvents. It also gives some of solvents properties and the solvent polarity function, f(ε, n2), values. The dependence of solvatochromic plots of the and on f(ε, n2) of 6AC in aprotic hydrogen-bond forming and in non-specifically interacting solvents are given in Figure 3. (circles) and (filled circles) as a function of f(ε, n2) (see Table 1 for definition) for 6AC in (1) acrylonitrile; (2) b propionitrile; (3) DBE; (4) THF; (5) DMF; (6) a DMSO; (7) HMPA; (8) a,b 1-chloro-n-hexadecane; (9) a,b 1-chloro-n-decane; (10) a,b 1-chloro-n-octane; (11) a,b 1-chloro-n-hexane; (12) a,b 1-chloro-n-butane; and (13) a,b 1-chloro-n-propane. a From reference [3], b From reference [4].
(circles) and (filled circles) as a function of f(ε, n2) (see Table 1 for definition) for 6AC in (1) acrylonitrile; (2) b propionitrile; (3) DBE; (4) THF; (5) DMF; (6) a DMSO; (7) HMPA; (8) a,b 1-chloro-n-hexadecane; (9) a,b 1-chloro-n-decane; (10) a,b 1-chloro-n-octane; (11) a,b 1-chloro-n-hexane; (12) a,b 1-chloro-n-butane; and (13) a,b 1-chloro-n-propane. a From reference [3], b From reference [4].
, in the spectra of 6AC in all solvents, which can form hydrogen bonds of acceptor character only, shift towards longer wavelength with respect to that in the spectrum of 6AC in 1-chloro-n-propane. The shapes of the long-wavelength band (Figure 2a) do not differ significantly in the spectra taken for all solvents used, but FWHM (full width at half maximum) in the absorption spectra, , increases slightly, while the batochromic shift increases significantly with increasing β Kamlet-Taft’s solvent parameter (Table 1). Similarly to all in the fluorescence spectra of 6AC in aprotic hydrogen-bond forming solvents (see Figure 2b and Table 1) are shifted towards longer wavelength with respect to that in the spectrum of 6AC in 1-chloro-n-propane. The shape of the fluorescence band (Figure 2b) and the FWHM value of the fluorescence spectra of 6AC, , are practically the same in all solvents used, similarly as that of the absorption spectra, but is lower than that for 6AC in 1-chloro-n-propane. No influence of the excitation wavelength, λexc, on the position and shape of the fluorescence spectrum was noted, similarly as reported earlier in references [4,5,39]. Very large Stokes shifts, in comparison with those of 7-aminocoumarins (e.g., [40,41]), have been observed between the absorption and fluorescence maxima (as reported earlier in reference [39]). This indicates that the changes in energy of solute–solvent specific (hydrogen bond formation) and non-specific interactions have a great influence on the static spectroscopic properties of 6AC. Table 1 lists the absorption and fluorescence maxima ( and , respectively), FWHM values of the absorption and fluorescence ( and , respectively) and the Stokes shifts, = − , of 6AC in different solvents. It also gives some of solvents properties and the solvent polarity function, f(ε, n2), values. The dependence of solvatochromic plots of the and on f(ε, n2) of 6AC in aprotic hydrogen-bond forming and in non-specifically interacting solvents are given in Figure 3. (circles) and (filled circles) as a function of f(ε, n2) (see Table 1 for definition) for 6AC in (1) acrylonitrile; (2) b propionitrile; (3) DBE; (4) THF; (5) DMF; (6) a DMSO; (7) HMPA; (8) a,b 1-chloro-n-hexadecane; (9) a,b 1-chloro-n-decane; (10) a,b 1-chloro-n-octane; (11) a,b 1-chloro-n-hexane; (12) a,b 1-chloro-n-butane; and (13) a,b 1-chloro-n-propane. a From reference [3], b From reference [4].
(circles) and (filled circles) as a function of f(ε, n2) (see Table 1 for definition) for 6AC in (1) acrylonitrile; (2) b propionitrile; (3) DBE; (4) THF; (5) DMF; (6) a DMSO; (7) HMPA; (8) a,b 1-chloro-n-hexadecane; (9) a,b 1-chloro-n-decane; (10) a,b 1-chloro-n-octane; (11) a,b 1-chloro-n-hexane; (12) a,b 1-chloro-n-butane; and (13) a,b 1-chloro-n-propane. a From reference [3], b From reference [4]. and , values for 6AC in aprotic hydrogen-bond forming solvents deviate from the linear correlations observed in 1-chloro-n-alkanes. This is a result of C type hydrogen bond formation between the solute and solvent molecules, which are stronger in the excited S1 state than in the ground S0 state [3]. Results of the theoretical study performed by Yang et al. [42] show that upon the S0→S1 excitation of the 6AC molecule, the electron density from the amino group in the ground S0 state disappears completely, and the electron density is localized over the entire molecule except the amino group. Therefore, the energies of hydrogen bonds formed by the solvent molecules with two hydrogen atoms of the amino group of 6AC should be significantly increased after excitation to the S1 state. Similarly to the 6AC molecule, the electron density redistribution from the amino group to the carbonyl group upon the S0→S1 excitation was found for the C120 molecule [36,43,44,45] and other coumarin molecules [46,47,48,49]. Moreover, on the basis of absorption and emission solvatochromic data obtained in this study (see Figure 3), as well as the results presented in references [4,5], it can be assumed that for 6AC and its complexes with solvent molecules, electron density on the nitrogen atom from the amino group decreases and that on the oxygen atom from carbonyl group increases in the emitting S1 state compared with those in the S1 state directly after excitation. or on the polarity function f(ε, n2) of the solvents used. The first step of the procedure required determination of the contribution coming from the nonspecific interactions only using the experimentally observed solvent spectral shifts in the absorption or emission spectra of the solute studied in the several 1-chloro-n-alkanes (interacting only non-specifically with solute). The evidence are the straight lines (see Figure 3) obtained as plots of the relation between or on the f(ε, n2). Therefore it can be assumed that these lines describe the effect of non-specific solute–solvent interactions on the and values not only in 1-chloro-n-alkanes but also in solvents that make hydrogen bonds with the solute, as long as these solvents satisfy the other assumptions of the Onsager reaction field model of interactions with the solute molecule. The distance between the straight line and the point corresponding to experimental or value in a given solvent is a measure of total ∆EHB of the hydrogen bonds formed between solute and solvent molecules. The values of ∆EHB experimentally obtained from spectral absorption and emission solvatochromic study (corresponding to S0→S1 and S1→S0 transitions, respectively) for 6AC-(solvent)n, n = 1, 2, complexes are collected in Table 2. The correlation between ∆EHB values and the Kamlet-Taft’s solvatochromic β solvent parameter are presented in Figure 4. As follows from Table 2 and Figure 4 for all 6AC-(solvent)n, n = 1, 2, complexes, the ∆EHB values obtained for S1→S0 emission process are higher than the corresponding ones for S0→S1 absorption process. The ∆EHB(em)/∆EHB(abs) ratio decreases almost linearly with increasing β Kamlet-Taft solvent parameter. The value of ∆EHB due to S0→S1 excitation process determined for 6AC-(DMSO)n complex is a bit lower than that estimated on the basis of the theoretical study for 6AC by Yang et al. [42]. The ∆EHB values obtained for 6AC-(DMSO)n and 6AC-(DMF)n complexes are also a bit lower than those calculated for C120-(DMSO)2 and C120-(DMF)2 complexes, respectively [36,45]. Unfortunately, there are no theoretical calculation data concerning S1→S0 emission process for intermolecular complexes of 6AC or other similar in the structure aminocoumarin derivatives with solvent.
and , values for 6AC in aprotic hydrogen-bond forming solvents deviate from the linear correlations observed in 1-chloro-n-alkanes. This is a result of C type hydrogen bond formation between the solute and solvent molecules, which are stronger in the excited S1 state than in the ground S0 state [3]. Results of the theoretical study performed by Yang et al. [42] show that upon the S0→S1 excitation of the 6AC molecule, the electron density from the amino group in the ground S0 state disappears completely, and the electron density is localized over the entire molecule except the amino group. Therefore, the energies of hydrogen bonds formed by the solvent molecules with two hydrogen atoms of the amino group of 6AC should be significantly increased after excitation to the S1 state. Similarly to the 6AC molecule, the electron density redistribution from the amino group to the carbonyl group upon the S0→S1 excitation was found for the C120 molecule [36,43,44,45] and other coumarin molecules [46,47,48,49]. Moreover, on the basis of absorption and emission solvatochromic data obtained in this study (see Figure 3), as well as the results presented in references [4,5], it can be assumed that for 6AC and its complexes with solvent molecules, electron density on the nitrogen atom from the amino group decreases and that on the oxygen atom from carbonyl group increases in the emitting S1 state compared with those in the S1 state directly after excitation. or on the polarity function f(ε, n2) of the solvents used. The first step of the procedure required determination of the contribution coming from the nonspecific interactions only using the experimentally observed solvent spectral shifts in the absorption or emission spectra of the solute studied in the several 1-chloro-n-alkanes (interacting only non-specifically with solute). The evidence are the straight lines (see Figure 3) obtained as plots of the relation between or on the f(ε, n2). Therefore it can be assumed that these lines describe the effect of non-specific solute–solvent interactions on the and values not only in 1-chloro-n-alkanes but also in solvents that make hydrogen bonds with the solute, as long as these solvents satisfy the other assumptions of the Onsager reaction field model of interactions with the solute molecule. The distance between the straight line and the point corresponding to experimental or value in a given solvent is a measure of total ∆EHB of the hydrogen bonds formed between solute and solvent molecules. The values of ∆EHB experimentally obtained from spectral absorption and emission solvatochromic study (corresponding to S0→S1 and S1→S0 transitions, respectively) for 6AC-(solvent)n, n = 1, 2, complexes are collected in Table 2. The correlation between ∆EHB values and the Kamlet-Taft’s solvatochromic β solvent parameter are presented in Figure 4. As follows from Table 2 and Figure 4 for all 6AC-(solvent)n, n = 1, 2, complexes, the ∆EHB values obtained for S1→S0 emission process are higher than the corresponding ones for S0→S1 absorption process. The ∆EHB(em)/∆EHB(abs) ratio decreases almost linearly with increasing β Kamlet-Taft solvent parameter. The value of ∆EHB due to S0→S1 excitation process determined for 6AC-(DMSO)n complex is a bit lower than that estimated on the basis of the theoretical study for 6AC by Yang et al. [42]. The ∆EHB values obtained for 6AC-(DMSO)n and 6AC-(DMF)n complexes are also a bit lower than those calculated for C120-(DMSO)2 and C120-(DMF)2 complexes, respectively [36,45]. Unfortunately, there are no theoretical calculation data concerning S1→S0 emission process for intermolecular complexes of 6AC or other similar in the structure aminocoumarin derivatives with solvent.| Solvent | ∆EHB | |
|---|---|---|
| S0→S1 | S1→S0 | |
| Acrylonitrile | 300 | 1420 |
| Propionitrile | 370 | 1040 |
| DBE | 300 | 1015 |
| THF | 580 | 930 |
| DMF | 1150 | 2120 |
| DMSO | 1340 a | 2530 |
| HMPA | 2000 | 2500 |

2.2. Hydrogen-Bonded Complexes in Ground S0 and Excited S1 States
2.3. Photophysical Study Results
energy gap for 6AC in (1) acrylonitrile, (2) propionitrile, (3) DBE, (4) THF, (5) DMF, (6) DMSO, (7) HMPA.
energy gap for 6AC in (1) acrylonitrile, (2) propionitrile, (3) DBE, (4) THF, (5) DMF, (6) DMSO, (7) HMPA.
 , at one wavelength from the short-wavelength side and another one from the long-wavelength side. For 6AC in acrylonitrile, propionitrile, DBE, DMF and DMSO, the fluorescence decays are single-exponential. In THF and HMPA the fluorescence decays of 6AC are found to follow non-single-exponential behaviour, and for them two-exponential function analysis gave reasonably good fits. For 6AC in THF the contribution of the long-time component (τ1) component was significantly greater than that of the short-time component (τ2). In HMPA two comparable time components (τ1,τ2) were found, but with significantly different contributions. The values of lifetime components for 6AC in all studied solvents are listed in Table 3. As the fluorescence decays of 6AC in aprotic hydrogen-bond forming solvents are usually single exponential, it is reasonable to assume that the fluorescence lifetimes of the 6AC-(solvent)1 and 6AC-(solvent)2 S1-excited complexes do not differ significantly in a particular solvent. Moreover, they must be similar to the fluorescence lifetime of 6AC in the S1-excited state in the same solvent, in analogy to 6AC in protic solvents [5] and to 4-aminophthalimide [1]. Because of the very high energy of hydrogen bonds formed by 6AC molecule with HMPA molecules in the S0 state, which are even strengthened due to S0→S1 excitation, there are practically only 6AC-(HMPA)2 S1-excited complexes present in HMPA solution in the S1-excited state. Therefore, the presence of two lifetime components in fluorescence decay must be associated with two types of emitting 6AC-(HMPA)2 complexes having different structures. Notably, these two lifetimes were found at each wavelength from the steady-state emission spectrum range, with the same wavelength independent contributions, which means that their presence in the 6AC decay is not a result of slow solvation.
, at one wavelength from the short-wavelength side and another one from the long-wavelength side. For 6AC in acrylonitrile, propionitrile, DBE, DMF and DMSO, the fluorescence decays are single-exponential. In THF and HMPA the fluorescence decays of 6AC are found to follow non-single-exponential behaviour, and for them two-exponential function analysis gave reasonably good fits. For 6AC in THF the contribution of the long-time component (τ1) component was significantly greater than that of the short-time component (τ2). In HMPA two comparable time components (τ1,τ2) were found, but with significantly different contributions. The values of lifetime components for 6AC in all studied solvents are listed in Table 3. As the fluorescence decays of 6AC in aprotic hydrogen-bond forming solvents are usually single exponential, it is reasonable to assume that the fluorescence lifetimes of the 6AC-(solvent)1 and 6AC-(solvent)2 S1-excited complexes do not differ significantly in a particular solvent. Moreover, they must be similar to the fluorescence lifetime of 6AC in the S1-excited state in the same solvent, in analogy to 6AC in protic solvents [5] and to 4-aminophthalimide [1]. Because of the very high energy of hydrogen bonds formed by 6AC molecule with HMPA molecules in the S0 state, which are even strengthened due to S0→S1 excitation, there are practically only 6AC-(HMPA)2 S1-excited complexes present in HMPA solution in the S1-excited state. Therefore, the presence of two lifetime components in fluorescence decay must be associated with two types of emitting 6AC-(HMPA)2 complexes having different structures. Notably, these two lifetimes were found at each wavelength from the steady-state emission spectrum range, with the same wavelength independent contributions, which means that their presence in the 6AC decay is not a result of slow solvation.| Solvent | ΦF a | λexc (nm) | τ1 (ps) | τ2 (ps) | kF 107 (s−1) | knr 107 (s−1) |
|---|---|---|---|---|---|---|
| 1-Chloro-n-propane b | 0.31 | 367 | 8200 | 3.78 | 8.4 | |
| Acrylonitrile | 0.18 | 370 | 9100 | 1.98 | 9.01 | |
| Propionitrile b | 0.26 | 380 | 12,580 | 2.07 | 5.88 | |
| DBE | 0.32 | 371 | 9050 | 3.53 | 7.51 | |
| THF | 0.30 | 374 | 13,500 (0.93) | 2700 (0.07) | 2.22 | 5.18 |
| DMF | 0.15 | 383 | 6610 | 2.27 | 12.8 | |
| DMSO | 0.084 | 387 | 4800 | 1.75 | 19.1 | |
| HMPA | 0.092 | 400 | 6000 (0.80) | 4000 (0.20) | 1.64 | 16.2 |
| Solvent | Mg→e (D) | Me→g (D) |
|---|---|---|
| Propionitrile a | 1.8 | 2.1 |
| DBE | 1.9 | 2.4 |
| THF | 1.9 | 2.0 |
| DMF | 1.9 | 2.2 |
| DMSO | 1.9 | 1.8 |
| HMPA | 1.9 | 1.8 |
 ;
;  ;
;  ; kF = ΦF/τF; ε(ν): molar extinction coefficient; n: refractive index; h: Planck constant; c: speed of light; NA: Avogadro constant; I(ν): fluorescence intensity at frequency ν; a From reference [4].
; kF = ΦF/τF; ε(ν): molar extinction coefficient; n: refractive index; h: Planck constant; c: speed of light; NA: Avogadro constant; I(ν): fluorescence intensity at frequency ν; a From reference [4].2.4. Deactivation of the Species Formed by 6AC in S1-Excited State in Aprotic Hydrogen-Bond Forming Solvents
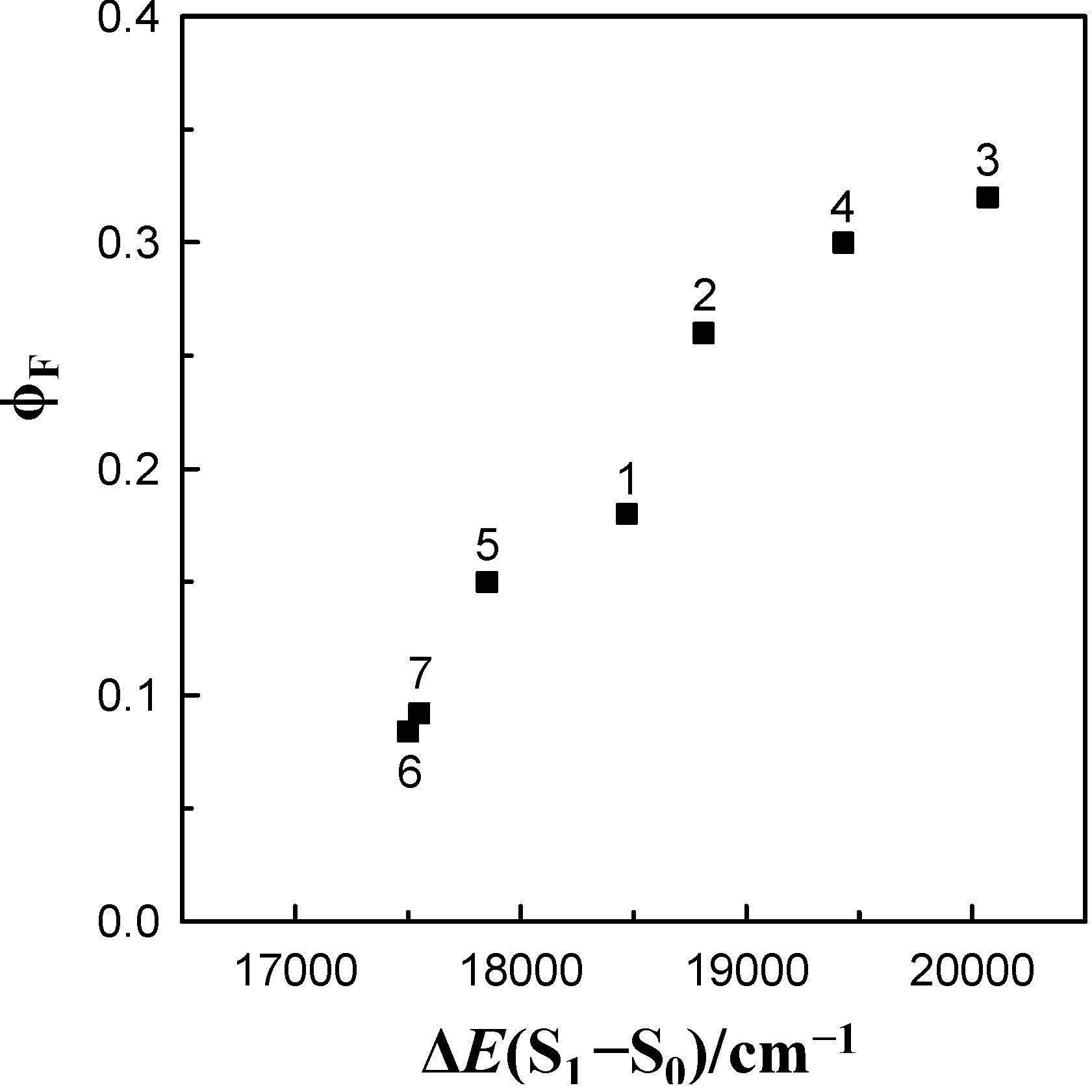
 energy gap. This figure demonstrates that the log knr value of these species for 6AC in aprotic hydrogen-bond forming solvents tends to increase linearly (except 6AC in DBE) with decreasing ∆E(S1–S0) and shows that the fast internal conversion is induced by the intermolecular solute-solvent hydrogen-bonding interactions, similarly as in protic solvents [5]. The nonradiative deactivation rate constant to the ground state is generally known to depend exponentially on the energy gap between the excited and ground states [60]. The results of this study, similarly to those in [5], clearly show that the energy gap dependence on radiationless deactivation in an internal conversion process from S1-excited state can be observed not only for molecules but also for hydrogen-bonded complexes. energy gap of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in (1) acrylonitrile, (2) propionitrile, (3) DBE, (4) THF, (5) DMF, (6) DMSO, (7) HMPA and of 6AC-(solvent)n, n = 2, 3, S1-excited complexes in (14) a 3,3,4,4,5,5,6,6,6-nonafluorohexanol, (15) a 1,1,1,3,3,3-hexafluoroisopropanol, (16) a 2,2,2-trifluoroethanol, (17) a H2O. a From reference [5].
energy gap of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in (1) acrylonitrile, (2) propionitrile, (3) DBE, (4) THF, (5) DMF, (6) DMSO, (7) HMPA and of 6AC-(solvent)n, n = 2, 3, S1-excited complexes in (14) a 3,3,4,4,5,5,6,6,6-nonafluorohexanol, (15) a 1,1,1,3,3,3-hexafluoroisopropanol, (16) a 2,2,2-trifluoroethanol, (17) a H2O. a From reference [5].
energy gap. This figure demonstrates that the log knr value of these species for 6AC in aprotic hydrogen-bond forming solvents tends to increase linearly (except 6AC in DBE) with decreasing ∆E(S1–S0) and shows that the fast internal conversion is induced by the intermolecular solute-solvent hydrogen-bonding interactions, similarly as in protic solvents [5]. The nonradiative deactivation rate constant to the ground state is generally known to depend exponentially on the energy gap between the excited and ground states [60]. The results of this study, similarly to those in [5], clearly show that the energy gap dependence on radiationless deactivation in an internal conversion process from S1-excited state can be observed not only for molecules but also for hydrogen-bonded complexes. energy gap of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in (1) acrylonitrile, (2) propionitrile, (3) DBE, (4) THF, (5) DMF, (6) DMSO, (7) HMPA and of 6AC-(solvent)n, n = 2, 3, S1-excited complexes in (14) a 3,3,4,4,5,5,6,6,6-nonafluorohexanol, (15) a 1,1,1,3,3,3-hexafluoroisopropanol, (16) a 2,2,2-trifluoroethanol, (17) a H2O. a From reference [5].
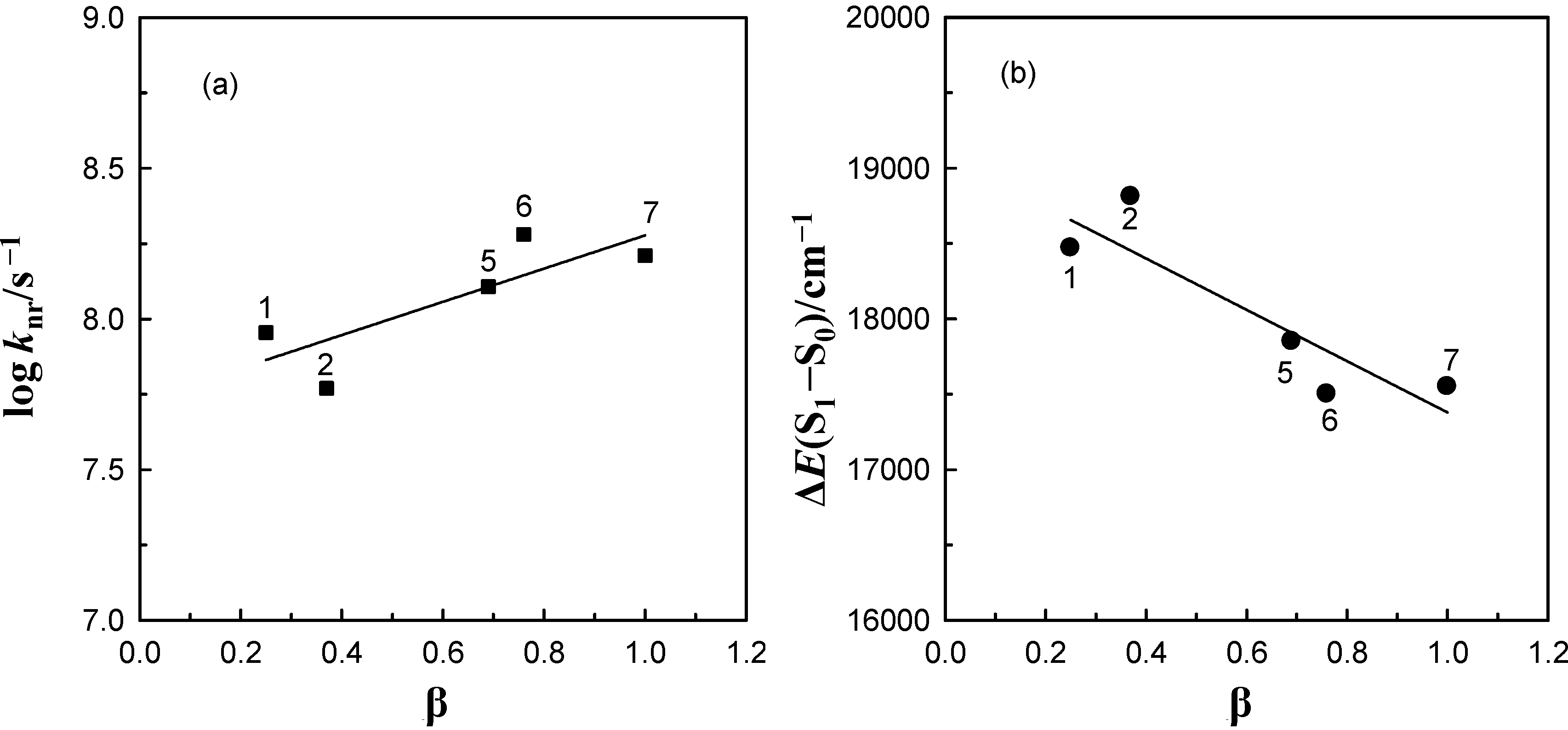
energy gap of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in (1) acrylonitrile, (2) propionitrile, (3) DBE, (4) THF, (5) DMF, (6) DMSO, (7) HMPA and of 6AC-(solvent)n, n = 2, 3, S1-excited complexes in (14) a 3,3,4,4,5,5,6,6,6-nonafluorohexanol, (15) a 1,1,1,3,3,3-hexafluoroisopropanol, (16) a 2,2,2-trifluoroethanol, (17) a H2O. a From reference [5]. energy gap (b) of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in acrylonitrile (1), propionitrile (2), DMF, (5), DMSO (6), HMPA (7) and the solvent Kamlet-Taft’s hydrogen-bonding acceptor parameter.
energy gap (b) of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in acrylonitrile (1), propionitrile (2), DMF, (5), DMSO (6), HMPA (7) and the solvent Kamlet-Taft’s hydrogen-bonding acceptor parameter.
energy gap (b) of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in acrylonitrile (1), propionitrile (2), DMF, (5), DMSO (6), HMPA (7) and the solvent Kamlet-Taft’s hydrogen-bonding acceptor parameter.
energy gap (b) of 6AC-(solvent)n, n = 1, 2, S1-excited complexes in acrylonitrile (1), propionitrile (2), DMF, (5), DMSO (6), HMPA (7) and the solvent Kamlet-Taft’s hydrogen-bonding acceptor parameter.
3. Experimental and Computational Methods
4. Conclusions
, is sensitive to solvent polarity, and similarly as the long wavelength absorption band maxima,  , to the hydrogen bonding ability. The photophysical study results clearly show that the intermolecular solute-solvent hydrogen bond formation, irrespective of the hydrogen bond character (donor and acceptor), induces an efficient radiationless deactivation of the S1-excited state through internal conversion. As shown for the first time in this study, for hydrogen-bonded complexes there is a linear dependence of the logarithm of the rate constant of nonradiative deactivation in an internal conversion process on the ∆E(S1–S0) energy-gap. Interestingly, the relationship between radiationless deactivation rate constant from S1-excited state and the energy gap in aprotic solvents was not much different from that in protic ones [5]. For 6AC in nonpolar aprotic solvents, besides fluorescence, efficient S1-ICT→S0 internal conversion arises from vibronic interactions between close-lying S1-ICT (π, π*) and S2 (n, π*) states.
, to the hydrogen bonding ability. The photophysical study results clearly show that the intermolecular solute-solvent hydrogen bond formation, irrespective of the hydrogen bond character (donor and acceptor), induces an efficient radiationless deactivation of the S1-excited state through internal conversion. As shown for the first time in this study, for hydrogen-bonded complexes there is a linear dependence of the logarithm of the rate constant of nonradiative deactivation in an internal conversion process on the ∆E(S1–S0) energy-gap. Interestingly, the relationship between radiationless deactivation rate constant from S1-excited state and the energy gap in aprotic solvents was not much different from that in protic ones [5]. For 6AC in nonpolar aprotic solvents, besides fluorescence, efficient S1-ICT→S0 internal conversion arises from vibronic interactions between close-lying S1-ICT (π, π*) and S2 (n, π*) states.Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krystkowiak, E.; Dobek, K.; Maciejewski, A. Origin of the strong effect of protic solvents on the emission spectra, quantum yield of fluorescence and fluorescence lifetime of 4-aminophthalimide: Role of hydrogen bonds in deactivation of S1-4-aminophthalimide. J. Photochem. Photobiol. A Chem. 2006, 184, 250–264. [Google Scholar]
- Krystkowiak, E.; Maciejewski, A.; Kubicki, J. Spectral and photophysical properties of thioxanthone in protic and aprotic solvents: The role of hydrogen bonds in S1-thioxanthone deactivation. ChemPhysChem 2006, 7, 597–606. [Google Scholar]
- Krystkowiak, E.; Maciejewski, A. Changes in energy of three types of hydrogen bonds upon excitation of aminocoumarins determined from absorption solvatochromic experiments. Phys. Chem. Chem. Phys. 2011, 13, 11317–11324. [Google Scholar]
- Krystkowiak, E.; Dobek, K.; Burdziński, G.; Maciejewski, A. Radiationless deactivation of 6-aminocoumarin from S1-ICT state in nonspecifically interacting solvents. Photochem. Photobiol. Sci. 2012, 11, 1322–1330. [Google Scholar]
- Krystkowiak, E.; Dobek, K.; Maciejewski, A. Intermolecular hydrogen-bonding effect on spectral and photophysical properties of 6-aminocoumarin in protic solvents. Photochem. Photobiol. Sci. 2013, 12, 446–455. [Google Scholar]
- Catalan, J. Toward a generalized treatment of the solvent effect based on four empirical scales: Dipolarity (SdP, a new scale), polarizability (SP), acidity (SA), and basicity (SB) of the medium. J. Phys. Chem. B 2009, 113, 5951–5960. [Google Scholar]
- Katritzky, A.R.; Fara, D.C.; Yang, H.; Tämm, K.; Tamm, T.; Karelson, M. Quantitative measures of solvent polarity. Chem. Rev. 2004, 104, 175–198. [Google Scholar]
- Kamlet, M.J.; Abboud, J.L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, pi*, α, and β, and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar]
- Marcus, Y.; Kamlet, M.J.; Taft, R.W. Linear solvation energy relationships: Standard molar Gibbs free energies and enthalpies of transfer of ions from water into nonaqueous solvents. J. Phys. Chem. 1988, 92, 3613–3622. [Google Scholar]
- Kamlet, M.J.; Taft, R.W. The solvatochromic comparison method. I. The β-scale of solvent hydrogen-bond acceptor (HBA) basicities. J. Am. Chem. Soc. 1976, 98, 377–383. [Google Scholar]
- Catalan, J.; Diaz, C.; Lopez, V.; Perez, P.; de Paz, J.-L.G.; Rodriguez, J.G. A generalized solvent basicity scale: The solvatochromism of 5-nitroindoline and its homomorph 1-methyl-5-nitroindoline. Liebigs Ann. 1996, 11, 1785–1794. [Google Scholar]
- Catalan, J. Acid-base interactions. In Handbook of Solvents; Wypych, G., Ed.; ChemTec Publishing William Andrew Publishing: Toronto, NY, USA, 2001; pp. 583–638. [Google Scholar]
- Marcus, Y. The Properties of Solvents; Wiley: Chichester, UK, 1998. [Google Scholar]
- Das, K.; Jain, B.; Patel, H.S. Hydrogen bonding properties of coumarin 151, 500, and 35: The effect of substitution at the 7-amino position. J. Phys. Chem. A 2006, 110, 1698–1704. [Google Scholar]
- Jones II, G.; Jackson, W.R.; Konaktanaporn, S. Solvent effects on photophysical parameters for coumarin laser dyes. Opt. Commun. 1980, 33, 315–320. [Google Scholar]
- Jones II, G.; Jackson, W.R.; Choi, C.Y.; Bergmark, W.R. Solvent effects on emission yield and lifetime for coumarin laser dyes: Requirements for a rotatory decay mechanism. J. Phys. Chem. 1985, 89, 294–300. [Google Scholar]
- Jones II, G.; Jackson, W.R.; Halpern, A.M. Medium effects on fluorescence quantum yields and lifetimes for coumarin laser dyes. Chem. Phys. Lett. 1980, 72, 391–395. [Google Scholar]
- Nandi, N.; Bhattacharyya, K.; Bagchi, B. Dielectric relaxation and solvation dynamics of water in complex chemical and biological systems. Chem. Rev. 2000, 100, 2013–2046. [Google Scholar]
- Demchenco, A.P. Advanced Fluorescence Reporters in Chemistry and Biology III: Applications in Sensing and Imaging; Springer: Berlin Heidelberg, Germany, 2011. [Google Scholar]
- Wagner, B.D. The use of coumarins as environmentally-sensitive fluorescent probes of heterogeneous inclusion systems. Molecules 2009, 14, 210–237. [Google Scholar]
- George, S.; Kumbhakar, M.; Singh, P.K.; Ganguly, R.; Nath, S.; Pal, H. Fluorescence spectroscopic investigation to identify the micelle to gel transition of aqueous triblock copolymer solutions. J. Phys. Chem. B 2009, 113, 5117–5127. [Google Scholar]
- Burai, T.N.; Datta, A. Slow solvation dynamics in the microheterogeneous water channels of nafion membranes. J. Phys. Chem. B 2009, 113, 15901–15906. [Google Scholar]
- Grazula, M.; Budzisz, E. Biological activity of metal ions complexes of chromones, coumarins and flavones. Coord. Chem. Rev. 2009, 253, 2588–2598. [Google Scholar]
- Oliveira, E.; Nunez, C.; Rodriguez-Gonzalez, B.; Capelo, J.L.; Lodeiro, C. Novel small stable gold nanoparticles bearing fluorescent cysteine-coumarin probes as new metal-modulated chemosensors. Inorg. Chem. 2011, 50, 8797–8807. [Google Scholar]
- Al Kady, A.S.; Gaber, M.; Hussein, M.M.; Ebeid, E.-Z.M. Fluorescence enhancement of coumarin thiourea derivatives by Hg2+, Ag+, and silver nanoparticles. J. Phys. Chem. A 2009, 113, 9474–9484. [Google Scholar]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar]
- Yu, H.; Li, J.; Wu, D.; Qiu, Z.; Zhang, Y. Chemistry and biological applications of photo-labile organic molecules. Chem. Soc. Rev. 2010, 39, 464–473. [Google Scholar]
- Kulkarni, M.V.; Kulkarni, G.M.; Lin, C.-H.; Sun, C.-M. Recent advances in coumarins and 1-azacoumarins as versatile biodynamic agents. Curr. Med. Chem. 2006, 13, 2795–2818. [Google Scholar]
- Madhavan, G.R.; Balraju, V.; Mallesham, B.; Chakrabarti, R.; Lohray, V.B. Novel coumarin derivatives of heterocyclic compounds as lipid-lowering agents. Bioorg. Med. Chem. Lett. 2003, 13, 2547–2551. [Google Scholar]
- Anufrik, S.S.; Tarkovsky, V.V. 3-(2-Benzimidazolyl)coumarin derivatives: Highly effective laser media. J. Appl. Spectr. 2010, 77, 640–647. [Google Scholar]
- Yang, Y.; Zou, J.; Rong, H.; Qian, G.D.; Wang, Z.Y.; Wang, M.Q. Influence of various coumarin dyes on the laser performance of laser dyes co-doped into ORMOSILs. Appl. Phys. B: Laser Opt. 2007, 86, 309–313. [Google Scholar]
- Nedumpara, R.J.; Thomas, K.J.; Jayasree, V.K.; Girijavallabhan, C.P.; Nampoori, V.P.N.; Radhakrishnan, P. Study of solvent effect in laser emission from coumarin 540 dye solution. Appl. Opt. 2007, 46, 4786–4792. [Google Scholar]
- Kopylova, T.N.; Mayer, G.V.; Reznichenko, A.V.; Samsonova, L.G.; Svetlichnyi, V.A.; Dolotov, S.M.; Ponomarenko, E.P.; Tavrizova, M.A. Solid-state active media based on aminocoumarins. Quantum Electron. 2003, 33, 498–502. [Google Scholar]
- Zhao, G.-J.; Han, K.-L. Early time hydrogen-bonding dynamics of photoexcited coumarin 102 in hydrogen-donating solvents: theoretical study. J. Phys. Chem. A 2007, 111, 2469–2474. [Google Scholar]
- Zhao, G.-J.; Han, K.-L. Hydrogen bonding in the electronic excited state. Acc. Chem. Res. 2012, 45, 404–413. [Google Scholar]
- Zhou, P.; Song, P.; Liu, J.; Han, K.; He, G. Experimental and theoretical study of the rotational reorientation dynamics of 7-aminocoumarin derivatives in polar solvents: Hydrogen-bonding effects. Phys. Chem. Chem. Phys. 2009, 11, 9440–9449. [Google Scholar]
- Liu, Y.; Ding, J.; Liu, R.; Shi, D.; Sun, J. Revisiting the electronic excited-state hydrogen bonding dynamics of coumarin chromophore in alcohols: Undoubtedly strengthened not cleaved. J. Photochem. Photobiol. A: Chem. 2009, 201, 203–207. [Google Scholar]
- Wells, N.P.; McGrath, M.J.; Siepmann, T.; Underwood, D.F.; Blanck, D.A. Excited state hydrogen bond dynamics: Coumarin 102 in acetonitrile-water binary mixtures. J. Phys. Chem. A 2008, 112, 2511–2514. [Google Scholar]
- Rettig, W.; Klock, A. Intramolecular fluorescence quenching in aminocoumarines: Identification of an excited state with full charge separation. Can. J. Chem. 1985, 63, 1649–1653. [Google Scholar]
- Nad, S.; Pal, H. Unusual photophysical properties of coumarin-151. J. Phys. Chem. A 2001, 105, 1097–1106. [Google Scholar]
- Pal, H.; Nad, S.; Kumbhakar, M. Photophysical properties of coumarin-120: Unusual behavior in nonpolar solvents. J. Chem. Phys. 2003, 119, 443–452. [Google Scholar]
- Yang, D.; Yang, Y.; Liu, Y. Effects of different-type intermolecular hydrogen bonds on the geometrical and spectral properties of 6-aminocoumarin clusters in solution. J. Clust. Sci. 2014, 25, 467–481. [Google Scholar]
- Xu, B.; Yang, J.; Jiang, X.; Wang, Y.; Sun, H.; Yin, J. Ground and excited calculations of 7-phenylamino-substituted coumarins. J. Mol. Struct. 2009, 917, 15–20. [Google Scholar]
- Han, K.-L.; Zhao, G.-J. Hydrogen Bonding and Transfer in the Excited State; John Wiley & Sons Ltd: Chichester, UK, 2011. [Google Scholar]
- Zhao, W.; Pan, L.; Bian, W.; Wang, J. Influence of solvent polarity and hydrogen bonding on the electronic transition of coumarin 120: A TDDFT study. ChemPhysChem 2008, 9, 1593–1602. [Google Scholar]
- Rechthaler, K.; Kohler, G. Excited state properties and deactivation pathways of 7-aminocoumarins. Chem. Phys. 1994, 189, 99–116. [Google Scholar]
- Samanta, A.; Fessenden, R.W. Excited-state dipole moment of 7-aminocoumarins as determined from time-resolved microwave dielectric absorption measurements. J. Phys. Chem. A 2000, 104, 8577–8582. [Google Scholar]
- Nemkovich, N.A.; Reis, H.; Baumann, W. Ground and excited state dipole moments of coumarin laser dyes: Investigation by electro-optical absorption and emission methods. J. Lumin. 1997, 71, 255–263. [Google Scholar]
- Aaron, J.-J.; Buna, M.; Parkanyi, C.; Antonious, M.S.; Tine, A.; Cisse, L. Quantitative treatment of the effect of solvent on the electronic absorption and fluorescence spectra of substituted coumarins: Evaluation of the first excited singlet-state dipole moments. J. Fluorescence 1995, 5, 337–347. [Google Scholar]
- Meech, S.R.; Phillips, D. Photophysics of some common fluorescence standards. J. Photochem. 1983, 23, 193–217. [Google Scholar]
- Birks, J.B. Photophysics of Aromatic Molecules; Wiley-Interscience: New York, NY, USA, 1970. [Google Scholar]
- Priyadarsini, K.I.; Naik, D.B.; Moorthy, P.N. A study of the triplet state of 7-amino coumarin laser dyes by the nanosecond pulse radiolysis technique. J. Photochem. Photobiol. A Chem. 1990, 54, 251–261. [Google Scholar]
- Jones II, G.; Jackson, W.R.; Kanoktanaporn, S.; Bergmark, W.R. Photophysical and photochemical properties of coumarin dyes in amphiphilic media. Photochem. Photobiol. 1985, 42, 477–483. [Google Scholar]
- Shimada, H.; Nakamura, A.; Yoshihara, T.; Tobita, S. Intramolecular and intermolecular hydrogen-bonding effects on photophysical properties of 2'-aminoacetophenone and its derivatives in solution. Photochem. Photobiol. Sci. 2005, 4, 367–375. [Google Scholar]
- Yoshihara, T.; Shimada, H.; Shizuka, H.; Tobita, S. Internal conversion of o-aminoacetophenone in solution. Phys. Chem. Chem. Phys. 2001, 3, 4972–4978. [Google Scholar]
- Zhao, G.-J.; Han, K.-L. Ultrafast hydrogen bond strengthening of the photoexcited fluorenone in alcohols for facilitating the fluorescence quenching. J. Phys. Chem. A 2007, 111, 9218–9223. [Google Scholar]
- Zhao, G.-J.; Han, K.-L. Role of intramolecular and intermolecular hydrogen bonding in both singlet and triplet excited states of aminofluorenones on internal conversion, intersystem crossing, and twisted intramolecular charge transfer. J. Phys. Chem. A 2009, 113, 14329–14335. [Google Scholar]
- Biczok, L.; Berces, T.; Yatsuhashi, T.; Tachibana, H.; Inoue, H. The role of intersystem crossing in the deactivation of the singlet excited aminofluorenones. Phys. Chem. Chem. Phys. 2001, 3, 980–985. [Google Scholar]
- Liu, Y.-H.; Zhao, G.-J.; Li, G.-Y.; Han, K.-L. Fluorescence quenching phenomena facilitated by excited-state hydrogen bond strengthening for fluorenone derivatives in alcohols. J. Photochem. Photobiol. A Chem. 2010, 209, 181–185. [Google Scholar]
- Englman, R.; Jortner, J. The energy gap law for radiationiess transitions in large molecules. Mol. Phys. 1970, 18, 145–164. [Google Scholar]
- Maciejewski, A.; Milewski, M.; Szymański, M. A method of determination of quantum yields of S3→S2, S3→S1, and S3→S0 intramolecular radiationless transitions. J. Chem. Phys. 1999, 111, 8462–8468. [Google Scholar]
- Medvedev, E.S.; Osherev, V.I. Radiationless Transitions in Polyatomic Molecules; Springer: Berlin, Germany, 1993. [Google Scholar]
- Freed, K.F. Radiationless Processes in Molecules and Condensed Phases; Fong, F. K., Ed.; Springer: Berlin, Germany, 1976; pp. 23–168. [Google Scholar]
- Morimoto, A.; Yatsuhashi, T.; Shimada, T.; Biczok, L.; Tryk, D.A.; Inoue, H. Radiationless deactivation of an intramolecular charge transfer excited state through hydrogen bonding: Effect of molecular structure and hard-soft anionic character in the excited state. J. Phys. Chem. A 2001, 105, 10488–10496. [Google Scholar]
- Karolczak, J.; Komar, D.; Kubicki, J.; Szymański, M.; Wróżowa, T.; Maciejewski, A. Fluorescence dynamics spectrometer of single-picosecond resolution: Optimisation of experimental performance. Bull. Pol. Acad. Sci. Chem. 1999, 47, 361–380. [Google Scholar]
- Karolczak, J.; Komar, D.; Kubicki, J.; Wróżowa, T.; Dobek, K.; Ciesielska, B.; Maciejewski, A. The measurements of picosecond fluorescence lifetimes with high accuracy and subpicosecond precision. Chem. Phys. Lett. 2001, 344, 154–164. [Google Scholar]
- Wróżowa, T.; Ciesielska, B.; Komar, D.; Karolczak, J.; Maciejewski, A.; Kubicki, J. Measurements of picosecond lifetimes by time correlated single photon counting method: The effect of the refraction index of the solvent on the instrument response function. Rev. Sci. Instrum. 2004, 75, 3107–3121. [Google Scholar]
- Krystkowiak, E.; Dobek, K.; Maciejewski, A. 6-Aminokumaryna–Sonda Do Badań Międzycząsteczkowych Wiązań Wodorowych w Roztworach; Nauka i przemysł–metody spektroskopowe w praktyce, nowe wyzwania i możliwości; Hubicki, Z., Ed.; Uniwersytet Marii Curie-Skłodowskiej w Lublinie, Wydział Chemii: Lublin, Poland, 2013; pp. 381–384. [Google Scholar]
- Krystkowiak, E.; Bachorz, R.A.; Koput, J. Ground and excited state hydrogen bonding affects of 6-aminocoumarin in water: An ab initio study. Dyes Pigment. 2014, 112, 335–340. [Google Scholar]
- Krystkowiak, E.; Koput, J.; Maciejewski, A. Hydrogen bond effects in the ground and excited singlet states of 4 H-1-benzopyrane-4-thione in water—Theory and experiment. Phys. Chem. Chem. Phys. 2012, 14, 8842–8851. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krystkowiak, E.; Dobek, K.; Maciejewski, A. Deactivation of 6-Aminocoumarin Intramolecular Charge Transfer Excited State through Hydrogen Bonding. Int. J. Mol. Sci. 2014, 15, 16628-16648. https://doi.org/10.3390/ijms150916628
Krystkowiak E, Dobek K, Maciejewski A. Deactivation of 6-Aminocoumarin Intramolecular Charge Transfer Excited State through Hydrogen Bonding. International Journal of Molecular Sciences. 2014; 15(9):16628-16648. https://doi.org/10.3390/ijms150916628
Chicago/Turabian StyleKrystkowiak, Ewa, Krzysztof Dobek, and Andrzej Maciejewski. 2014. "Deactivation of 6-Aminocoumarin Intramolecular Charge Transfer Excited State through Hydrogen Bonding" International Journal of Molecular Sciences 15, no. 9: 16628-16648. https://doi.org/10.3390/ijms150916628
APA StyleKrystkowiak, E., Dobek, K., & Maciejewski, A. (2014). Deactivation of 6-Aminocoumarin Intramolecular Charge Transfer Excited State through Hydrogen Bonding. International Journal of Molecular Sciences, 15(9), 16628-16648. https://doi.org/10.3390/ijms150916628